
Abstract
Arrhythmogenic right ventricular dysplasia/cardiomyopathy is an inherited cardiomyopathy estimated to affect approximately 1 in 5,000 individuals. Cardinal manifestations include right ventricular enlargement and dysfunction, fibrofatty replacement of myocytes in the right ventricle, characteristic electrocardiographic abnormalities, and ventricular arrhythmia most commonly arising from the right ventricle. The disease is frequently familial and typically involves autosomal dominant transmission with low penetrance and variable expressivity. Approximately 50% of symptomatic individuals harbor a mutation in one of the five major components of the cardiac desmosome. Nevertheless, other genetic modifiers and environmental factors complicate the clinical management of mutation carriers as well as counseling of their relatives. This Review summarizes the known genetic mutations associated with arrhythmogenic right ventricular dysplasia/cardiomyopathy, describes possible origins of recurrent mutations, presents theories on the pathogenesis of disease following a mutation, and discusses the current issues surrounding clinical use of genetic analysis in the assessment of individuals with this condition.
Key Points
-
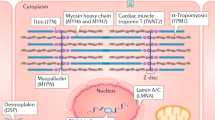
Mutation in genes encoding any of the five major components of the cardiac desmosome—PKP2 (encoding plakophilin-2), DSG2 (encoding desmoglein-2), DSP (encoding desmoplakin), DSC2 (encoding desmocollin-2), and JUP (encoding junctional plakoglobin)—can result in arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C)
-
Approximately 50% of individuals with ARVD/C who have undergone full sequence analysis of these desmosome genes have a single heterozygous mutation identified, though a few cases of individuals with homozygous or compound heterozygous mutations have also been described
-
ARVD/C segregates in families with both incomplete penetrance and variable expressivity; clinical screening of family members is recommended, particularly among those recognized to share a genetic predisposition to ARVD/C
-
Owing to the age-dependent onset of ARVD/C, repeat clinical screening is recommended at 2- to 3-year intervals from the age of 12 years in the absence of a known mutation, to help target family members at highest risk; in families with earlier onset disease or sudden cardiac death in children, earlier clinical screening should be performed
-
With the recent emergence of clinical genetic testing for ARVD/C, genetic counseling is strongly advised for individuals with ARVD/C and their family members
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


References
McKenna WJ et al. (1994) Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J 71: 215–218
Uhl HS (1952) A previously undescribed congenital malformation of the heart: almost total absence of the myocardium of the right ventricle. Bull Johns Hopkins Hosp 91: 197–209
Gerlis LM et al. (1993) Dysplastic conditions of the right ventricular myocardium: Uhl's anomaly vs arrhythmogenic right ventricular dysplasia. Br Heart J 69: 142–150
Frank R et al. (1978) Electrocardiology of 4 cases of right ventricular dysplasia inducing arrhythmia [French]. Arch Mal Coeur Vaiss 71: 963–972
Nava A et al. (1987) A polymorphic form of familial arrhythmogenic right ventricular dysplasia. Am J Cardiol 59: 1405–1409
Nava A et al. (1988) Familial occurrence of right ventricular dysplasia: a study involving nine families. J Am Coll Cardiol 12: 1222–1228
Marcus FI et al. (1982) Right ventricular dysplasia: a report of 24 adult cases. Circulation 65: 384–398
Coonar AS et al. (1998) Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation 97: 2049–2058
McKoy G et al. (2000) Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355: 2119–2124
Protonotarios N et al. (2001) Genotype–phenotype assessment in autosomal recessive arrhythmogenic right ventricular cardiomyopathy (Naxos disease) caused by a deletion in plakoglobin. J Am Coll Cardiol 38: 1477–1484
Asimaki A et al. (2007) A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 81: 964–973
Getsios S et al. (2004) Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol 5: 271–281
Bierkamp C et al. (1996) Embryonic heart and skin defects in mice lacking plakoglobin. Dev Biol 180: 780–785
Ruiz P et al. (1996) Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol 135: 215–225
Kirchhof P et al. (2006) Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation 114: 1799–1806
Rao BH et al. (1996) Familial occurrence of a rare combination of dilated cardiomyopathy with palmoplantar keratoderma and curly hair. Indian Heart J 48: 161–162
Carvajal-Huerta L (1998) Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J Am Acad Dermatol 39: 418–421
Norgett EE et al. (2000) Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 9: 2761–2766
Alcalai R et al. (2003) A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair. J Am Coll Cardiol 42: 319–327
Norgett EE . et al. (2006) Early death from cardiomyopathy in a family with autosomal dominant striate palmoplantar keratoderma and woolly hair associated with a novel insertion mutation in desmoplakin. 126: 1651–1654
Uzumcu A et al. (2006) Loss of desmoplakin isoform I causes early onset cardiomyopathy and heart failure in a Naxos-like syndrome. J Med Genet 43: e5
Bauce B et al. (2005) Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J 26: 1666–1675
Rampazzo A et al. (2002) Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 71: 1200–1206
Norman M et al. (2005) Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation 112: 636–642
Jonkman MF et al. (2005) Loss of desmoplakin tail causes lethal acantholytic epidermolysis bullosa. Am J Hum Genet 77: 653–660
Whittock NV et al. (2002) Compound heterozygosity for non-sense and mis-sense mutations in desmoplakin underlies skin fragility/woolly hair syndrome. J Invest Dermatol 118: 232–238
Smith EA and Fuchs E (1998) Defining the interactions between intermediate filaments and desmosomes. J Cell Biol 141: 1229–1241
Gallicano GI et al. (1998) Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J Cell Biol 143: 2009–2022
Gallicano GI et al. (2001) Rescuing desmoplakin function in extra-embryonic ectoderm reveals the importance of this protein in embryonic heart, neuroepithelium, skin and vasculature. Development 128: 929–941
Garcia-Gras E et al. (2006) Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest 116: 2012–2021
Yang Z et al. (2006) Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res 99: 646–655
Grossmann KS et al. (2004) Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol 167: 149–160
Gerull B et al. (2004) Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet 36: 1162–1164
Dalal D et al. (2006) Clinical features of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in plakophilin-2. Circulation 113: 1641–1649
Syrris P et al. (2006) Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation 113: 356–364
van Tintelen JP et al. (2006) Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 113: 1650–1658
Awad MM et al. (2006) Recessive arrhythmogenic right ventricular dysplasia due to novel cryptic splice mutation in PKP2. Hum Mutat 27: 1157
Lahtinen AM et al. (2007) Plakophilin-2 missense mutations in arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol [10.1016/j.ijcard.2007.03.137]
Dalal D et al. (2006) Penetrance of mutations in plakophilin-2 among families with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol 48: 1416–1424
Nava A et al. (2000) Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 36: 2226–2233
Hamid MS et al. (2002) Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol 40: 1445–1450
Bonne S et al. (2000) Assignment of the plakophilin-2 gene (PKP2) and a plakophilin-2 pseudogene (PKP2P1) to human chromosome bands 12p11 and 12p13, respectively, by in situ hybridization. Cytogenet Cell Genet 88: 286–287
Schwarz MA et al. (1990) Desmosomes and hemidesmosomes: constitutive molecular components. Annu Rev Cell Biol 6: 461–491
Awad MM et al. (2006) DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Hum Genet 79: 136–142
Pilichou K et al. (2006) Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 113: 1171–1179
Syrris P et al. (2007) Desmoglein-2 mutations in arrhythmogenic right ventricular cardiomyopathy: a genotype-phenotype characterization of familial disease. Eur Heart J 28: 581–588
Sen-Chowdhry S et al. (2007) Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 115: 1710–1720
Eshkind L et al. (2002) Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur J Cell Biol 81: 592–598
Biedermann K et al. (2005) Desmoglein 2 is expressed abnormally rather than mutated in familial and sporadic gastric cancer. J Pathol 207: 199–206
Yashiro M et al. (2006) Decreased expression of the adhesion molecule desmoglein-2 is associated with diffuse-type gastric carcinoma. Eur J Cancer 42: 2397–2403
Heuser A et al. (2006) Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 79: 1081–1088
Syrris P et al. (2006) Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet 79: 978–984
Beffagna G et al. (2007) Missense mutations in desmocollin-2 N-terminus, associated with arrhythmogenic right ventricular cardiomyopathy, affect intracellular localization of desmocollin-2 in vitro. BMC Med Genet 8: 65
Priori SG et al. (2001) Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103: 196–200
Tiso N et al. (2001) Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 10: 189–194
Rampazzo A et al. (1995) A new locus for arrhythmogenic right ventricular cardiomyopathy (ARVD2) maps to chromosome 1q42-q43. Hum Mol Genet 4: 2151–2154
Beffagna G et al. (2005) Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res 65: 366–373
Ahmad F et al. (1998) Localization of a gene responsible for arrhythmogenic right ventricular dysplasia to chromosome 3p23. Circulation 98: 2791–2795
Li D et al. (2000) The locus of a novel gene responsible for arrhythmogenic right-ventricular dysplasia characterized by early onset and high penetrance maps to chromosome 10p12-p14. Am J Hum Genet 66: 148–156
Rampazzo A et al. (1997) ARVD4, a new locus for arrhythmogenic right ventricular cardiomyopathy, maps to chromosome 2 long arm. Genomics 45: 259–263
Severini GM et al. (1996) A new locus for arrhythmogenic right ventricular dysplasia on the long arm of chromosome 14. Genomics 31: 193–200
Hodgkinson KA et al. (2005) The impact of implantable cardioverter-defibrillator therapy on survival in autosomal-dominant arrhythmogenic right ventricular cardiomyopathy (ARVD5). J Am Coll Cardiol 45: 400–408
Simcha I et al. (1998) Differential nuclear translocation and transactivation potential of beta-catenin and plakoglobin. J Cell Biol 141: 1433–1448
Ross SE et al. (2000) Inhibition of adipogenesis by Wnt signaling. Science 289: 950–953
Nagata M et al. (2000) Apoptotic cell death in arrhythmogenic right ventricular cardiomyopathy: a comparative study with idiopathic sustained ventricular tachycardia. Jpn Heart J 41: 733–741
Yamaji K et al. (2005) Apoptotic myocardial cell death in the setting of arrhythmogenic right ventricular cardiomyopathy. Acta Cardiol 60: 465–470
Mallat Z et al. (1996) Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N Engl J Med 335: 1190–1196
Hakimelahi S et al. (2000) Plakoglobin regulates the expression of the anti-apoptotic protein BCL-2. J Biol Chem 275: 10905–10911
Longo KA et al. (2002) Wnt signaling protects 3T3-L1 preadipocytes from apoptosis through induction of insulin-like growth factors. J Biol Chem 277: 38239–38244
Shaw RM et al. (2007) Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 128: 547–560
Oxford EM et al. (2007) Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ Res 101: 703–711
Saffitz JE et al. (2007) Remodeling of gap junctions in ischemic and nonischemic forms of heart disease. J Membr Biol 218: 65–71
Acknowledgements
The authors wish to acknowledge funding from the National Institutes of Health (HL088072 to DPJ) and the France-Merrick Foundation. We would also like to acknowledge the Johns Hopkins ARVD Program (http://www.arvd.com) which is supported by the Bogle Foundation, the Campanella family, and the Wilmerding Endowments. Désirée Lie, University of California, Irvine, CA, is the author of and is solely responsible for the content of the learning objectives, questions and answers of the Medscape-accredited continuing medical education activity associated with this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Awad, M., Calkins, H. & Judge, D. Mechanisms of Disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Rev Cardiol 5, 258–267 (2008). https://doi.org/10.1038/ncpcardio1182
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ncpcardio1182
This article is cited by
-
A novel DSP zebrafish model reveals training- and drug-induced modulation of arrhythmogenic cardiomyopathy phenotypes
Cell Death Discovery (2023)
-
Accurate Classification of Non-ischemic Cardiomyopathy
Current Cardiology Reports (2023)
-
Impaired Plakophilin-2 in obesity breaks cell cycle dynamics to breed adipocyte senescence
Nature Communications (2023)
-
Epidemiology of the inherited cardiomyopathies
Nature Reviews Cardiology (2021)
-
“Ryanopathies” and RyR2 dysfunctions: can we further decipher them using in vitro human disease models?
Cell Death & Disease (2021)
