
Key Points
-
Neuromuscular disorders comprise a heterogeneous group of clinical conditions that primarily affect one or more components of the neuromuscular unit — typically skeletal muscle — but are often also multisystemic. The considerable clinical impact of these diseases is exemplified by the muscular dystrophies and by spinal muscular atrophy (SMA), for which the development for novel molecular therapies is both urgent and challenging.
-
Recent advances in RNA biology have accelerated the progress of a new generation of molecular therapies based on RNA. Single- or double-stranded nucleic acid agents and small-molecule agents are being developed as novel therapies to target the mutant mRNAs that are involved in neuromuscular disease. These approaches modulate pre-mRNA processing or inhibit the deleterious effects of toxic RNAs.
-
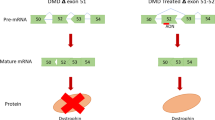
In the case of Duchenne muscular dystrophy (DMD), exon-skipping therapies to restore a viable open reading frame in the DMD gene are well advanced. Two recent systemic delivery clinical trials have reported encouraging data on the restoration of dystrophin protein expression, and second-generation compounds are in development. An alternative approach to stimulate the readthrough of premature stop codons, which is applicable to a subset of patients with DMD, is also in development.
-
Myotonic dystrophy arises as a result of an expanded microsatellite repeat mutation that leads to a gain-of-function toxic mRNA, which ultimately causes muscle degeneration and multisystem dysfunction. Various approaches to correct the deleterious toxic effects of toxic RNA using small-molecule-based, oligonucleotide-based or RNA interference-based methods to inhibit or degrade the toxic RNA are being investigated.
-
A viable therapeutic strategy for SMA, which arises as a result of the loss-of-function of the survival of motor neuron 1 (SMN1) gene, is to restore the splicing of exon 7 in the related SMN2 gene, which has the potential to fully compensate for the loss of the SMN1 protein. Recent studies using oligonucleotide-mediated exon-inclusion methods appear to be highly promising.
-
Although the development of RNA-based therapies for neuromuscular disease remains challenging, recent progress in this field is encouraging. However, major barriers remain the poor in vivo delivery of most RNA therapeutic agents and the regulatory hurdles that are associated with the development of novel personalized medicines.
Abstract
The development of effective therapies for neuromuscular disorders such as Duchenne muscular dystrophy (DMD) is hampered by considerable challenges: skeletal muscle is the most abundant tissue in the body, and many neuromuscular disorders are multisystemic conditions. However, despite these barriers there has recently been substantial progress in the search for novel treatments. In particular, the use of antisense oligonucleotides, which are designed to target RNA and modulate pre-mRNA splicing to restore functional protein isoforms or directly inhibit the toxic effects of pathogenic RNAs, offers great promise and these approaches are now being tested in the clinic. Here, we review recent advances in the development of such antisense oligonucleotides and other promising novel approaches, including the induction of readthrough nonsense mutations.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


References
Davies, K. E. & Nowak, K. J. Molecular mechanisms of muscular dystrophies: old and new players. Nature Rev. Mol. Cell Biol. 7, 762–773 (2006).
Bushby, K. et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 9, 177–189 (2010).
Bushby, K. et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 9, 77–93 (2010).
Muntoni, F., Torelli, S. & Ferlini, A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2, 731–740 (2003).
Manzur, A. Y., Kinali, M. & Muntoni, F. Update on the management of Duchenne muscular dystrophy. Arch. Dis. Child. 93, 986–990 (2008).
Emery, A. & Muntoni, F. (eds) Duchenne Muscular Dystrophy (Oxford University Press, New York, 2003).
Hoffman, E. P., Brown, R. H., Jr & Kunkel, L. M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928 (1987).
Hoffman, E. P., Monaco, A. P., Feener, C. C. & Kunkel, L. M. Conservation of the Duchenne muscular dystrophy gene in mice and humans. Science 238, 347–350 (1987).
Osborne, R. J. & Thornton, C. A. RNA-dominant diseases. Hum. Mol. Genet. 2006 (Suppl. 2), R162–R169 (2006).
Ranum, L. P. & Cooper, T. A. RNA-mediated neuromuscular disorders. Annu. Rev. Neurosci. 29, 259–277 (2006).
Wheeler, T. M. & Thornton, C. A. Myotonic dystrophy: RNA-mediated muscle disease. Curr. Opin. Neurol. 20, 572–576 (2007).
Burghes, A. H. & Beattie, C. E. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nature Rev. Neurosci. 10, 597–609 (2009).
Talbot, K. & Davies, K. E. Spinal muscular atrophy. Semin. Neurol. 21, 189–197 (2001).
Zhang, Z. et al. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133, 585–600 (2008).
Stephenson, M. L. & Zamecnik, P. C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl Acad. Sci. USA 75, 285–288 (1978).
Marwick, C. First “antisense” drug will treat CMV retinitis. JAMA 280, 871 (1998).
Kurreck, J. Antisense technologies. Improvement through novel chemical modifications. Eur. J. Biochem. 270, 1628–1644 (2003).
Lebedeva, I. & Stein, C. A. Antisense oligonucleotides: promise and reality. Annu. Rev. Pharmacol. Toxicol. 41, 403–419 (2001).
Opalinska, J. B. & Gewirtz, A. M. Nucleic-acid therapeutics: basic principles and recent applications. Nature Rev. Drug Discov. 1, 503–514 (2002).
Bennett, C. F. & Swayze, E. E. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 50, 259–293 (2010).
Dominski, Z. & Kole, R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl Acad. Sci. USA 90, 8673–8677 (1993).
Ahmad, A., Brinson, M., Hodges, B. L., Chamberlain, J. S. & Amalfitano, A. Mdx mice inducibly expressing dystrophin provide insights into the potential of gene therapy for duchenne muscular dystrophy. Hum. Mol. Genet. 9, 2507–2515 (2000).
Ghahramani Seno, M. M. et al. RNAi-mediated knockdown of dystrophin expression in adult mice does not lead to overt muscular dystrophy pathology. Hum. Mol. Genet. 17, 2622–2632 (2008).
Bushby, K. M. & Gardner-Medwin, D. The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. I. Natural history. J. Neurol. 240, 98–104 (1993).
Khurana, T. S. & Davies, K. E. Pharmacological strategies for muscular dystrophy. Nature Rev. Drug Discov. 2, 379–390 (2003).
Chamberlain, J. S. Stem-cell biology: a move in the right direction. Nature 444, 552–553 (2006).
Wang, Z. et al. Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol. Ther. 15, 1160–1166 (2007).
Mendell, J. R. et al. Dystrophin immunity in Duchenne's muscular dystrophy. N. Engl. J. Med. 363, 1429–1437 (2010). This study reported the detection of dystrophin-specific T cells targeting truncated dystrophin epitopes following the delivery of a functional dystrophin transgene in patients with DMD.
Berry, S. E., Liu, J., Chaney, E. J. & Kaufman, S. J. Multipotential mesoangioblast stem cell therapy in the mdx/utrn−/− mouse model for Duchenne muscular dystrophy. Regen. Med. 2, 275–288 (2007).
Galvez, B. G. et al. Complete repair of dystrophic skeletal muscle by mesoangioblasts with enhanced migration ability. J. Cell Biol. 174, 231–243 (2006).
Dellavalle, A. et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nature Cell Biol. 9, 255–267 (2007).
van Deutekom, J. C. & van Ommen, G. J. Advances in Duchenne muscular dystrophy gene therapy. Nature Rev. Genet. 4, 774–783 (2003).
Grange, R. W., Gainer, T. G., Marschner, K. M., Talmadge, R. J. & Stull, J. T. Fast-twitch skeletal muscles of dystrophic mouse pups are resistant to injury from acute mechanical stress. Am. J. Physiol. Cell Physiol. 283, C1090–C1101 (2002).
Dunckley, M. G., Manoharan, M., Villiet, P., Eperon, I. C. & Dickson, G. Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Hum. Mol. Genet. 7, 1083–1090 (1998).
Pramono, Z. A. et al. Induction of exon skipping of the dystrophin transcript in lymphoblastoid cells by transfecting an antisense oligodeoxynucleotide complementary to an exon recognition sequence. Biochem. Biophys. Res. Commun. 226, 445–449 (1996).
Takeshima, Y., Nishio, H., Sakamoto, H., Nakamura, H. & Matsuo, M. Modulation of in vitro splicing of the upstream intron by modifying an intra-exon sequence which is deleted from the dystrophin gene in dystrophin Kobe. J. Clin. Invest. 95, 515–520 (1995).
Aartsma-Rus, A. et al. Targeted exon skipping as a potential gene correction therapy for Duchenne muscular dystrophy. Neuromuscul. Disord. 12 (Suppl. 1), 71–77 (2002).
Aartsma-Rus, A. et al. Comparative analysis of antisense oligonucleotide analogs for targeted DMD exon 46 skipping in muscle cells. Gene Ther. 11, 1391–1398 (2004).
Popplewell, L. J. et al. Comparative analysis of antisense oligonucleotide sequences targeting exon 53 of the human DMD gene: implications for future clinical trials. Neuromuscul. Disord. 20, 102–110 (2010).
Popplewell, L. J., Trollet, C., Dickson, G. & Graham, I. R. Design of phosphorodiamidate morpholino oligomers (PMOs) for the induction of exon skipping of the human DMD gene. Mol. Ther. 17, 554–561 (2009).
van Deutekom, J. C. et al. Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum. Mol. Genet. 10, 1547–1554 (2001).
Bulfield, G., Siller, W. G., Wight, P. A. & Moore, K. J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl Acad. Sci. USA 81, 1189–1192 (1984).
Lu, Q. L. et al. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nature Med. 9, 1009–1014 (2003). This was the first in vivo study reporting the restoration of functional dystrophin protein following the intramuscular administration of an antisense oligonucleotide in the mdx mouse.
Mann, C. J. et al. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc. Natl Acad. Sci. USA 98, 42–47 (2001).
Mann, C. J., Honeyman, K., McClorey, G., Fletcher, S. & Wilton, S. D. Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J. Gene Med. 4, 644–654 (2002).
Sazani, P. et al. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nature Biotech. 20, 1228–1233 (2002).
Lu, Q. L. et al. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl Acad. Sci. USA 102, 198–203 (2005). This was the first in vivo study reporting the successful restoration of dystrophin protein in multiple peripheral muscle groups following the systemic intravenous administration of an antisense oligonucleotide in the mdx mouse.
Heemskerk, H. et al. Preclinical PK and PD studies on 2′-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol. Ther. 18, 1210–1217 (2010).
Alter, J. et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nature Med. 12, 175–177 (2006).
Yokota, T. et al. Efficacy of systemic morpholino exon-skipping in duchenne dystrophy dogs. Ann. Neurol. 65, 667–676 (2009).
Malerba, A. et al. Chronic systemic therapy with low-dose morpholino oligomers ameliorates the pathology and normalizes locomotor behavior in mdx mice. Mol. Ther. 19, 345–354 (2011).
Malerba, A., Thorogood, F. C., Dickson, G. & Graham, I. R. Dosing regimen has a significant impact on the efficiency of morpholino oligomer-induced exon skipping in mdx mice. Hum. Gene Ther. 20, 955–965 (2009).
Wu, B. et al. Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Ther. 17, 132–140 (2010).
Fayssoil, A., Nardi, O., Orlikowski, D. & Annane, D. Cardiomyopathy in Duchenne muscular dystrophy: pathogenesis and therapeutics. Heart Fail. Rev. 15, 103–107 (2010).
Heemskerk, H. A. et al. In vivo comparison of 2′-O-methyl phosphorothioate and morpholino antisense oligonucleotides for Duchenne muscular dystrophy exon skipping. J. Gene Med. 11, 257–266 (2009).
Levin, A. A. A review of the issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim. Biophys. Acta 1489, 69–84 (1999).
Henry, S. P. et al. Complement activation is responsible for acute toxicities in rhesus monkeys treated with a phosphorothioate oligodeoxynucleotide. Int. Immunopharmacol. 2, 1657–1666 (2002).
Wang, Q. et al. In vitro evaluation of novel antisense oligonucleotides is predictive of in vivo exon skipping activity for Duchenne muscular dystrophy. J. Gene Med. 12, 354–364 (2010).
Crisp, A. et al. Diaphragm rescue alone prevents heart dysfunction in dystrophic mice. Hum. Mol. Genet. 20, 413–421 (2011).
Muntoni, F. et al. Brief report: deletion of the dystrophin muscle-promoter region associated with X-linked dilated cardiomyopathy. N. Engl. J. Med. 329, 921–925 (1993).
Townsend, D., Yasuda, S., Li, S., Chamberlain, J. S. & Metzger, J. M. Emergent dilated cardiomyopathy caused by targeted repair of dystrophic skeletal muscle. Mol. Ther. 16, 832–835 (2008).
McClorey, G., Moulton, H. M., Iversen, P. L., Fletcher, S. & Wilton, S. D. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 13, 1373–1381 (2006).
Nelson, M. H. et al. Arginine-rich peptide conjugation to morpholino oligomers: effects on antisense activity and specificity. Bioconjug. Chem. 16, 959–966 (2005).
Abes, S. et al. Peptide-based delivery of nucleic acids: design, mechanism of uptake and applications to splice-correcting oligonucleotides. Biochem. Soc. Trans. 35, 53–55 (2007).
Abes, S. et al. Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents. J. Control Release 116, 304–313 (2006).
Amantana, A. et al. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug. Chem. 18, 1325–1331 (2007).
Fletcher, S. et al. Morpholino oligomer-mediated exon skipping averts the onset of dystrophic pathology in the mdx mouse. Mol. Ther. 15, 1587–1592 (2007).
Jearawiriyapaisarn, N. et al. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol. Ther. 16, 1624–1629 (2008).
Wu, B. et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc. Natl Acad. Sci. USA 105, 14814–14819 (2008).
Yin, H., Lu, Q. & Wood, M. Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol. Ther. 16, 38–45 (2008). The studies in Refs 68–70 reported a considerably enhanced restoration of dystrophin protein following the systemic administration of a peptide-conjugated antisense oligonucleotide.
Moulton, H. M. et al. Cell-penetrating peptide-morpholino conjugates alter pre-mRNA splicing of DMD (Duchenne muscular dystrophy) and inhibit murine coronavirus replication in vivo. Biochem. Soc. Trans. 35, 826–828 (2007).
Moulton, H. M. et al. Peptide-morpholino conjugate: a promising therapeutic for Duchenne muscular dystrophy. Ann. NY Acad. Sci. 1175, 55–60 (2009).
Samoylova, T. I. & Smith, B. F. Elucidation of muscle-binding peptides by phage display screening. Muscle Nerve 22, 460–466 (1999).
Yin, H., Moulton, H., Betts, C. & Wood, M. CPP-directed oligonucleotide exon skipping in animal models of Duchenne muscular dystrophy. Methods Mol. Biol. 683, 321–338 (2011).
Yin, H. et al. Functional rescue of dystrophin-deficient mdx mice by a chimeric peptide-PMO. Mol. Ther. 18, 1822–1829 (2010).
Yin, H. et al. A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum. Mol. Genet. 18, 4405–4414 (2009).
Yin, H. et al. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol. Ther. 19, 1295–1303 (2011).
Ivanova, G. D. et al. Improved cell-penetrating peptide–PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 36, 6418–6428 (2008).
Ivanova, G. D. et al. PNA-peptide conjugates as intracellular gene control agents. Nucleic Acids Symp. Ser. (Oxf.) 52, 31–32 (2008).
Amantana, A. & Iversen, P. L. Pharmacokinetics and biodistribution of phosphorodiamidate morpholino antisense oligomers. Curr. Opin. Pharmacol. 5, 550–555 (2005).
Iversen, P. L. Phosphorodiamidate morpholino oligomers: favorable properties for sequence-specific gene inactivation. Curr. Opin. Mol. Ther. 3, 235–238 (2001).
Crooke, S. T. et al. Pharmacokinetic properties of several novel oligonucleotide analogs in mice. J. Pharmacol. Exp. Ther. 277, 923–937 (1996).
Morandi, L. et al. Dystrophin characterization in BMD patients: correlation of abnormal protein with clinical phenotype. J. Neurol. Sci. 132, 146–155 (1995).
Kinali, M. et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 8, 918–928 (2009).
van Deutekom, J. C. et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 357, 2677–2686 (2007). This was the first clinical study to report the successful restoration of dystrophin protein following the intramuscular administration of a 2OMe antisense oligonucleotide in patients with DMD.
Aartsma-Rus, A. Antisense-mediated modulation of splicing: therapeutic implications for Duchenne muscular dystrophy. RNA Biol. 7, 453–461 (2010).
Aartsma-Rus, A. et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 30, 293–299 (2009).
Helderman-van den Enden, A. T. et al. Becker muscular dystrophy patients with deletions around exon 51; a promising outlook for exon skipping therapy in Duchenne patients. Neuromuscul. Disord. 20, 251–254 (2010).
Furling, D. et al. Viral vector producing antisense RNA restores myotonic dystrophy myoblast functions. Gene Ther. 10, 795–802 (2003).
Saengpattrachai, M., Ray, P. N., Hawkins, C. E., Berzen, A. & Banwell, B. L. Grandpa and I have dystrophinopathy?: approach to asymptomatic hyperCKemia. Pediatr. Neurol. 35, 145–149 (2006).
Goemans, N. M. et al. Systemic administration of PRO051 in duchenne's muscular dystrophy. N. Engl. J. Med. 364, 1513–1522 (2011). This was another clinical study that reported the successful restoration of dystrophin protein following systemic administration of a 2OMe antisense oligonucleotide in patients with DMD.
Wagner, K. R. et al. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann. Neurol. 49, 706–711 (2001).
Dunant, P., Walter, M. C., Karpati, G. & Lochmuller, H. Gentamicin fails to increase dystrophin expression in dystrophin-deficient muscle. Muscle Nerve 27, 624–627 (2003).
Barton-Davis, E. R., Cordier, L., Shoturma, D. I., Leland, S. E. & Sweeney, H. L. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Invest. 104, 375–381 (1999).
Malik, V. et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann. Neurol. 67, 771–780 (2010).
Welch, E. M. et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 447, 87–91 (2007). This study identified a chemical entity, PTC124, that selectively induced ribosomal readthrough of premature termination codons and demonstrated efficacy in promoting dystrophin protein production in primary human muscle cells and in mdx mice.
Howard, M. T. et al. Readthrough of dystrophin stop codon mutations induced by aminoglycosides. Ann. Neurol. 55, 422–426 (2004).
Day, J. W. et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology 60, 657–664 (2003).
Jiang, H., Mankodi, A., Swanson, M. S., Moxley, R. T. & Thornton, C. A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 13, 3079–3088 (2004).
Lee, J. E. & Cooper, T. A. Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 37, 1281–1286 (2009).
Mahadevan, M. S. et al. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nature Genet. 38, 1066–1070 (2006).
Ranum, L. P. & Day, J. W. Myotonic dystrophy: RNA pathogenesis comes into focus. Am. J. Hum. Genet. 74, 793–804 (2004).
Orengo, J. P. et al. Expanded CTG repeats within the DMPK 3′ UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc. Natl Acad. Sci. USA 105, 2646–2651 (2008).
Ward, A. J., Rimer, M., Killian, J. M., Dowling, J. J. & Cooper, T. A. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum. Mol. Genet. 19, 3614–3622 (2010).
Yadava, R. S. et al. RNA toxicity in myotonic muscular dystrophy induces NKX2-5 expression. Nature Genet. 40, 61–68 (2008).
Yuan, Y. et al. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res. 35, 5474–5486 (2007).
Liquori, C. L. et al. Myotonic dystrophy type 2: human founder haplotype and evolutionary conservation of the repeat tract. Am. J. Hum. Genet. 73, 849–862 (2003).
Margolis, J. M., Schoser, B. G., Moseley, M. L., Day, J. W. & Ranum, L. P. DM2 intronic expansions: evidence for CCUG accumulation without flanking sequence or effects on ZNF9 mRNA processing or protein expression. Hum. Mol. Genet. 15, 1808–1815 (2006).
Cooper, T. A. Molecular biology. Neutralizing toxic RNA. Science 325, 272–273 (2009).
Koshelev, M., Sarma, S., Price, R. E., Wehrens, X. H. & Cooper, T. A. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum. Mol. Genet. 19, 1066–1075 (2010).
Orengo, J. P., Ward, A. J. & Cooper, T. A. Alternative splicing dysregulation secondary to skeletal muscle regeneration. Ann. Neurol. 69, 681–690 (2010).
Ho, T. H. et al. Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J. Cell Sci. 118, 2923–2933 (2005).
Lin, X. et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 15, 2087–2097 (2006).
Kuyumcu-Martinez, N. M., Wang, G. S. & Cooper, T. A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell 28, 68–78 (2007).
Wang, G. S., Kearney, D. L., De Biasi, M., Taffet, G. & Cooper, T. A. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J. Clin. Invest. 117, 2802–2811 (2007).
Osborne, R. J. et al. Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum. Mol. Genet. 18, 1471–1481 (2009).
Wang, G. S. et al. PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J. Clin. Invest. 119, 3797–3806 (2009).
Ranum, L. P. & Day, J. W. Pathogenic RNA repeats: an expanding role in genetic disease. Trends Genet. 20, 506–512 (2004).
Du, H. et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nature Struct. Mol. Biol. 17, 187–193 (2010).
Ho, T. H., Bundman, D., Armstrong, D. L. & Cooper, T. A. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum. Mol. Genet. 14, 1539–1547 (2005).
Mulders, S. A., van Engelen, B. G., Wieringa, B. & Wansink, D. G. Molecular therapy in myotonic dystrophy: focus on RNA gain-of-function. Hum. Mol. Genet. 19, R90–R97 (2010).
Cooper, T. A. Chemical reversal of the RNA gain of function in myotonic dystrophy. Proc. Natl Acad. Sci. USA 106, 18433–18434 (2009).
Warf, M. B., Nakamori, M., Matthys, C. M., Thornton, C. A. & Berglund, J. A. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl Acad. Sci. USA 106, 18551–18556 (2009). This study identified the ability of the small molecule pentamidine to reverse missplicing of pre-mRNAs that are implicated in myotonic dystrophy.
Wheeler, T. M. et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 325, 336–339 (2009). This study showed the reversal of a transgenic myotonic dystrophy phenotype using a PMO antisense oligonucleotide, CAG25, that binds to microsatellite expanded RNA and blocks its interaction with MBNL1.
Mulders, S. A. et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl Acad. Sci. USA 106, 13915–13920 (2009).
Krol, J. et al. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol. Cell 25, 575–586 (2007). This was a report on the use of a 2OMe (CAG) 7 antisense oligonucleotide that silences RNA expression of mutated DMPK , reduces the number of ribonuclear aggregates and normalizes aberrant pre-mRNA splicing in vivo in mouse models of DM1.
Langlois, M. A. et al. Cytoplasmic and nuclear retained DMPK mRNAs are targets for RNA interference in myotonic dystrophy cells. J. Biol. Chem. 280, 16949–16954 (2005).
Passini, M. A. & Cheng, S. H. Prospects for the gene therapy of spinal muscular atrophy. Trends Mol. Med. 17, 259–265 (2011).
Foust, K. D. et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nature Biotech. 28, 271–274 (2010).
Valori, C. F. et al. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2, 35ra42 (2010).
Wheeler, T. M., Lueck, J. D., Swanson, M. S., Dirksen, R. T. & Thornton, C. A. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J. Clin. Invest. 117, 3952–3957 (2007).
Passini, M. A. et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 3, 72ra18 (2011). This study reported the use of methoxyethyl-modified antisense oligonucleotides that were administered directly to the CNS in mice and cynomolgus monkeys, and demonstrated successful splicing redirection of the SMN2 gene.
Politano, L. et al. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol. 22, 15–21 (2003).
Baughan, T. et al. Stimulating full-length SMN2 expression by delivering bifunctional RNAs via a viral vector. Mol. Ther. 14, 54–62 (2006).
Baughan, T. D., Dickson, A., Osman, E. Y. & Lorson, C. L. Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum. Mol. Genet. 18, 1600–1611 (2009).
Dickson, A., Osman, E. & Lorson, C. L. A negatively acting bifunctional RNA increases survival motor neuron both in vitro and in vivo. Hum. Gene Ther. 19, 1307–1315 (2008).
Skordis, L. A., Dunckley, M. G., Yue, B., Eperon, I. C. & Muntoni, F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc. Natl Acad. Sci. USA 100, 4114–4119 (2003).
Hua, Y. et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 24, 1634–1644 (2010). This was the first study to demonstrate effective in vivo CNS splicing redirection of the SMN2 gene in the CNS in a mouse model of SMA.
Hua, Y., Vickers, T. A., Baker, B. F., Bennett, C. F. & Krainer, A. R. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 5, e73 (2007).
Hua, Y., Vickers, T. A., Okunola, H. L., Bennett, C. F. & Krainer, A. R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 82, 834–848 (2008).
Owen, N. et al. Design principles for bifunctional targeted oligonucleotide enhancers of splicing. Nucleic Acids Res. 20 May 2011 (doi:10.1093/nar/gkr152).
Burghes, A. H. & McGovern, V. L. Antisense oligonucleotides and spinal muscular atrophy: skipping along. Genes Dev. 24, 1574–1579 (2010).
Cartegni, L. & Krainer, A. R. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nature Struct. Biol. 10, 120–125 (2003).
Coady, T. H., Shababi, M., Tullis, G. E. & Lorson, C. L. Restoration of SMN function: delivery of a trans-splicing RNA re-directs SMN2 pre-mRNA splicing. Mol. Ther. 15, 1471–1478 (2007).
Singh, N. N., Shishimorova, M., Cao, L. C., Gangwani, L. & Singh, R. N. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 6, 341–350 (2009).
Meyer, K. et al. Rescue of a severe mouse model for spinal muscular atrophy by U7 snRNA-mediated splicing modulation. Hum. Mol. Genet. 18, 546–555 (2009).
Alvarez-Erviti, L. et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nature Biotech. 29, 341–345 (2011).
Mitrpant, C. et al. Rational design of antisense oligomers to induce dystrophin exon skipping. Mol. Ther. 17, 1418–1426 (2009).
Muntoni, F. The development of antisense oligonucleotide therapies for Duchenne muscular dystrophy: report on a TREAT-NMD workshop hosted by the European Medicines Agency (EMA), on September 25th 2009. Neuromuscul. Disord. 20, 355–362 (2010).
Lee, J. E. & Cooper, T. A. Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 37, 1281–1286 (2009).
Yin, H., Lu, Q. & Wood, M. Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol. Ther. 16, 38–45 (2008).
Yin, H. et al. Optimization of peptide nucleic acid antisense oligonucleotides for local and systemic dystrophin splice correction in the mdx mouse. Mol. Ther. 18, 819–827 (2010).
Quinlivan, R., Kirschner, J., Comi, G. & Bushby, K. for the Ataluren DBMD Study Group. Safety and efficacy of ataluren 10,10,20 mg/kg TID in patients with nonsense mutation dystrophinopathy. European Paediatric Neurology Society Congress, 9th EPNS Congress [online], (2011).
Acknowledgements
F.M. is supported by the Great Ormond Street Children's Charity. The financial support of the Medical Research Council (MRC), the Muscular Dystrophy Campaign, the French Muscular Dystrophy Association (AFM), the Wellcome Trust and TREAT-NMD is also gratefully acknowledged. M.J.A.W. is supported by the MRC, the AFM, the Wellcome Trust, Parkinson's UK, Action Duchenne, the Muscular Dystrophy Campaign, Duchenne Ireland and Muscular Dystrophy Ireland. The authors wish to thank Q. Lu, Neuromuscular/ALS Center, Carolinas Medical Center, Charlotte, North Carolina, USA, for providing Figure 2.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Francesco Muntoni has previously served on the scientific advisory boards for AVI BioPharma, he has a patent pending regarding tailed antisense oligonucleotides to redirect splicing, and his university receives research support from AVI BioPharma and GlaxoSmithKline in relation to clinical trials on antisense oligonucleotides.
Matthew J. A. Wood has a patent pending regarding peptides for the delivery of antisense oligonucleotides.
Related links
Related links
FURTHER INFORMATION
Francesco Muntoni's homepages
Centre for Neuromuscular Diseases
Glossary
- Neuromuscular disease
-
A disease affecting one or more constituent components of the neuromuscular unit, which comprise motor neurons, neuromuscular synapses and skeletal muscle.
- Duchenne muscular dystrophy
-
(DMD). A severe, X-linked neuromuscular disease that arises as a result of mutations in the dystrophin gene that abolish the open reading frame, leading to the absence of dystrophin protein.
- Myotonic dystrophy
-
The most common cause of muscular dystrophy in adults, which arises as a result of microsatellite repeat expansion mutations in the myotonin protein kinase gene (leading to muscular dystrophy type 1) or the cellular nucleic acid-binding protein gene (leading to muscular dystrophy type 2). These mutations lead to the formation of directly pathogenic RNAs and widespread dysregulation of splicing.
- Spinal muscular atrophy
-
(SMA). A very common childhood genetic disease that arises as a result of loss-of-function mutations in the survival of motor neuron 1 gene, which lead to motor neuron degeneration and subsequent axial and limb muscle weakness.
- Pre-mRNA splicing
-
The processing of a pre-mRNA transcript to remove specific intronic and some exonic sequences that are not required for the generation of the mature mRNA.
- RNA-targeted therapies
-
Therapeutic modalities that are based on the targeted modification or inhibition of RNAs (for example, pre-mRNA, mature mRNA or non-coding RNA) that are directly implicated in disease.
- Antisense oligonucleotide
-
A short single-stranded nucleic acid, typically 15–25 nucleotides in length, that has the ability to mediate therapeutic effects by directly interacting with pre-mRNA or mRNA in a sequence-specific manner.
- 2′-O-methyl RNA
-
An antisense oligonucleotide in which the RNA backbone is modified by the addition of methyl (CH3) groups at the 2′ position in the ribose ring.
- Phosphorodiamidate morpholino
-
(PMO). An extensively modified antisense oligonucleotide that is neutrally charged and comprises a morpholino ring-based RNA backbone.
- Open reading frame
-
(ORF). A stretch of a DNA sequence that does not contain a stop or termination codon, and typically corresponds to the legible DNA sequence of a single gene.
- Exon skipping
-
The processing of a pre-mRNA transcript to result in the exclusion of a specific exon within the mature mRNA transcript.
- mdx mouse
-
A naturally occurring mouse mutant that carries a premature termination codon in exon 23 of the dystrophin (Dmd) gene, which results in the absence of dystrophin protein.
- Premature termination codon
-
A mutation that results in the presence of a de novo termination or stop codon in advance of where one would normally be found, and which serves to disrupt the open reading frame for that gene.
- Cell-penetrating peptides
-
Short peptide fragments that are able to mediate enhanced cellular uptake of a range of cargoes by virtue of their cationic charge and the presence of specific cationic amino acid residues, typically arginine residues.
- (RXR)4
-
An arginine-rich cell-penetrating peptide comprising eight arginine residues and the non-natural amino acid X (6-aminohexanoic acid).
- Peptide–PMO
-
(PPMO). A phosphorodiamidate morpholino (PMO) oligonucleotide to which a short peptide fragment is attached at either the 5′ or 3′ end by direct chemical conjugation.
- Stop codon readthrough
-
A therapeutic method of addressing the deleterious effect of a premature termination codon in which the readthrough of a premature termination codon is stimulated in order to yield a full-length open reading frame.
- Microsatellite repeat expansion
-
The presence of repeat units of a microsatellite sequence in the genome, which expand beyond the number of repeat units that are associated with normal health, thus leading to a mutation that is directly pathogenic.
- Spliceopathy
-
A disease that arises through widespread dysregulation of pre-mRNA splicing.
- Exon inclusion
-
The processing of a pre-mRNA transcript to result in the inclusion of a specific exon within the mature mRNA transcript.
- Personalized molecular therapies
-
The development of therapeutic agents that are directly tailored to specific mutations or to individual patients or subsets of patients.
Rights and permissions
About this article
Cite this article
Muntoni, F., Wood, M. Targeting RNA to treat neuromuscular disease. Nat Rev Drug Discov 10, 621–637 (2011). https://doi.org/10.1038/nrd3459
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrd3459
This article is cited by
-
Tritium labeling of antisense oligonucleotides via different conjugation agents
AAPS Open (2021)
-
LINC00673 is activated by YY1 and promotes the proliferation of breast cancer cells via the miR-515-5p/MARK4/Hippo signaling pathway
Journal of Experimental & Clinical Cancer Research (2019)
-
A novel antisense oligonucleotide anchored on the intronic splicing enhancer of hTERT pre-mRNA inhibits telomerase activity and induces apoptosis in glioma cells
Journal of Neuro-Oncology (2019)
-
Injection site reactions after long-term subcutaneous delivery of drisapersen: a retrospective study
European Journal of Pediatrics (2019)
-
Antisense oligonucleotides: the next frontier for treatment of neurological disorders
Nature Reviews Neurology (2018)
