Article Text

Abstract
Objective: To characterise and investigate the functional consequences of a novel TNFRSF1A splice site mutation causing tumour necrosis factor receptor associated periodic syndrome (TRAPS) in a 16-year-old male patient and his mother.
Methods: Mutational DNA screening was performed in the patient and his mother. Western blotting was used to analyse protein expression levels of TNFR1. A multiplex bead immunoassay was used to quantify serum levels of range of cytokines, and an ELISA-based transcription factor assay to measure nuclear factor (NF)-κB transactivation. Serum levels of soluble TNFR1 (sTNFR1) were measured by ELISA and fluorescence-activated cell sorting (FACS) analysis used to measure monocyte TNFR1 cell surface expression.
Results: A novel mutation, c.472+1G>A (C158delinsYERSSPEAKPSPHPRG), involving a splice site in intron 4 of TNFRSF1A, was found in the proband and affected mother leading to a 45 nucleotide insertion of intronic DNA into the mRNA, resulting in an in-frame insertion of 15 amino acids in the mature TNFR1 protein and a deletion of a cysteine residue C129 (158) in cysteine rich domain (CRD)3. The patients had reduced serum sTNFR1 and surface expression levels of TNFR1, with marked increases in pro- and anti-inflammatory cytokine. Their peripheral blood mononuclear cells (PBMC) had increased basal NF-κB activation compared with healthy controls and also had increased p50 nuclear expression following tumour necrosis factor (TNF) stimulation compared with PBMC from healthy controls, as well as T50M (T79M) and C88R (C117R) patients with TRAPS and patients with rheumatoid arthritis (RA).
Conclusion: A novel, TRAPS causing, TNFRSF1A splice site mutation is associated with decreased sTNFR1 levels, cell surface and whole cell extract expression and increased NF-κB transcription factor activation.
Statistics from Altmetric.com
Tumour necrosis factor receptor associated periodic syndrome (TRAPS) is an autosomal dominant autoinflammatory disorder caused by mutations in the TNFRSF1A gene,1–9 characterised by recurrent attacks of fever, abdominal pain, synovial inflammation, conjunctivitis, periorbital oedema and a positive family history in most cases. The TNFRSF1A gene comprises 10 exons and translates to a 455 amino acid precursor, where 29 amino acids are cleaved from the amino terminus to give the mature TNFR1 protein. More than 50 different pathogenic mutations have been described (INFEVERS database, http://fmf.igh.cnrs.fr/infevers).10 11 The majority (over 90%) of TNFRSF1A mutations are single nucleotide missense mutations within exons 2, 3, 4 and 6; however, one mutation, creating a splice site in intron 2 and two deletions, c.211_213del5 and c.293_295del, has also been reported (INFEVERS database). Most mutations described to date are located in the extracellular region in cysteine rich domains, with the single exception of I170N (I199N) (the codon number in the parenthesis refers to the conventional codon number including the 29 amino acid leader sequence) located close to the receptor cleavage site, between Asn172 (201) and Val173 (202).12
The mechanisms by which mutations in the TNFRSF1A gene cause the clinical features of this disease are still largely unknown. Several independent groups have proposed multiple mechanisms, including impaired shedding,1 abnormal apoptosis,13 and defective trafficking.14 15 Several reports have relied upon expression models with immortalised cell lines or the transfection of truncated mutant TNFR1 constructs to study the cellular mechanisms of this disease. This has lead to several contradictory findings that are likely to be more reflective of the model rather than the particular mutation involved. Furthermore, it is emerging that there are likely to be mutation specific mechanisms at work that are responsible for the wide range in disease severity observed, as demonstrated by low penetrance mutations, such as P46L (P75L) and R92Q (R121Q), and the more severe pathology associated with mutations involving cysteine residues and T50 (T79) mutations.3 5
Here we describe a novel mutation, involving a splice site in intron 4 of TNFRSF1A, in a 16-year-old male patient with a long-standing history of multiple previously unexplained episodic symptoms, including recurrent fevers and abdominal pain. Previous investigations had excluded inflammatory bowel disease, familial Mediterranean fever (FMF) and hyper IgD and periodic fever syndrome (HIDS). Using primary cells, we also report that this TNFRSF1A mutation leads to increased nuclear factor κappaB (NF-κB) activation.
MATERIALS AND METHODS
Patients and health controls
The study was approved by the Leeds (East) Research Ethics Committee. Blood samples were obtained from patients and healthy controls, all with informed consent. Disease control groups for the functional studies were two patients with TRAPS with known mutations T50M (T79M) and C88R (C117R) and three patients with rheumatoid arthritis (RA). The peripheral blood mononuclear cells (PBMC) were isolated by gradient lymphoprep (Invitrogen, Paisley, UK) centrifugation, counted and then divided for cryopreservation, fluorescence-activated cell sorting (FACS) analysis, RNA and protein extraction.
PCR and DNA sequencing
DNA was isolated from whole blood using a DNeasy spin column (Qiagen, Crawley, UK) and PCR was carried out as described previously.1 Primers designed for cDNA PCR were TNFR 3-6 F: GCTCCAAATGCCGAAAGG and TNFR 3-6 R CGTGCACTCCAGGCTTTTCT (MWG Biotech, Ebersberg, Germany). PCR conditions were as described previously.1
The products were purified using ExoSAP (USB, Staufen, Germany) and prepared for sequencing using BigDye 3.1 chemistry. Sequencing was carried out on the ABI3130 Genetic Analyser (Applied Biosystems, Foster City, California, USA). The cDNA PCR products were cloned into pGEM-T-Easy (ProMega, Southampton, UK) and sequenced as above.
Quantitative real-time PCR
RNA was isolated from the PBMC using a phenol/chloroform extraction method and cDNA reverse transcribed using Superscript II (Invitrogen). Quantitative real-time PCR was carried out on AB7900, using Applied Biosystems core kit reagents for 50 cycles. Exon targeting primers were TNFR F: GTGCTTCAATTGCAGCCTCTG and TNFR R: CCTGCATGGCAGGTGCA (GenBank accession no. TNFRSF1A: NM_001065). Product size was determined using 2% Tris/acetate/EDTA (TAE) agarose gel electrophoresis.
Western blot analysis for TNFR1 expression
Protein lysates were prepared from isolated PBMC using a cytsolic/nuclear fractionation kit (Biovision, Palo Alto, California, USA). Lysate were normalised to protein and subjected to 10% sodium dodecylsulphate (SDS)-polyacrylamide gel electrophoresis (PAGE) under reducing conditions and analysed by Western blotting (WB). Blots were probed with a goat polyclonal anti-human TNFR1 antibody, raised against an epitope mapping at the N-terminus (clone N-20; SCBT, Santa Cruz, California, USA), and bound immunoglobulin was detected with an enhanced chemiluminescence kit (GE Healthcare, Amersham, UK); blots were subsequently re-probed with anti-β actin monoclonal antibody (MoAb) (Sigma Aldrich, Gillingham, UK), as a loading control. Protein bands were quantified using BioRad Quantity One 4.6 software (Bio Rad, Hemel Hempstead, UK) and expressed as previously a ratio (TNFR1/β-actin).
Measurement of sTNFR1 levels in sera
The concentrations of sTNFR1 were measured using a commercially available ELISA assay (R&D Systems, Abingdon, UK), as previously described.1
TNFR1 expression analysis by FACS analysis
The surface and intracellular expression of TNFR1 was studied by FACS analysis in monocytes from the proband and his mother. PBMC were isolated from blood using Lymphoprep (Nycomed Pharma AS, Zurich, Switzerland) and stained with FITC-labelled TNFR1 MoAb (Serotec, Oxford, UK). To identify the monocyte population, double staining was carried out by simultaneous incubation with a phycoerythrin (PE)-labelled or fluorescein isothiocyanate (FITC)-labelled antibody respectively, recognising the monocyte marker CD14 (BD Bioscience, Oxford, UK). Corresponding isotype control MoAbs (BD Bioscience), were used as negative controls. For analysis of intracellular TNFR1 expression the cells were first fixed and permeabilised (Caltag, supplied by Invitragen) prior to staining. TNFR1 shedding was measured following PBMC stimulation with 50 ng/ml phorbal 12-myristate 13-acetate PMA (Sigma, UK)/250 ng/ml ionomycin (Sigma) for 4 h. Cells were acquired into a FACScan flow cytometer (BD Bioscience). Monocytes were gated by their forward/side scatter profiles and CD14 expression.
NF-κB transactivation analysis
Nuclear proteins were extracted using the cytosolic/nuclear fractionation kit (Biovision), and quantified using the BCA method. Transactivation of NF-κB p50 and p65 subunits was determined using a p50/p65 NF-κB ELISA-based transcription factor assay kit (TransAM assay; Active Motif Europe, Rixensart, Belgium). Absorbance at 450 nm was read on an OpsysMR microplate reader (Dynex Technologies, Eaton Socon, UK).
Cytokine quantification
Serum cytokine levels of tumour necrosis factor (TNF), interleukin (IL)1β, IL6, IL10, IL2 and IL12 were determined, using a multiplex bead immunoassay kit (Biosource, supplied by Invitrogen). Following the last wash step, the beads were resuspended in buffer and analysed in duplicate with a Luminex 100 instrument (Luminex, Riverside, California, USA). Data analysis was performed using the Luminex 100 IS software version 2.3 and results displayed as mean (SD).
RESULTS
Novel TNFRSF1A splice site mutation in proband and mother with TRAPS
The proband, a 16-year-old male, was well until aged 3 years when he presented with a fever of 4 days duration, abdominal pain and diarrhoea. He had markedly raised inflammatory markers and a white blood cell (WBC) count of 15.2×109/litre, with neutrophilia of 11.9×109/litre and C-reactive protein (CRP) of 265 mg/litre. A year later he had developed sore throat, headache, generalised non-migratory erythaematosus rash and fever, and was treated as presumed tonsillitis. Over the following years he had several admissions with episodes of generalised abdominal pain, sometimes accompanied by vomiting and diarrhoea. Between these episodes he was well and developed steadily along the 25th percentile for height and weight. From year 2000 he had recurrent episodic fevers, lymphangitis and pleuritis with groin and musculoskeletal pains affecting his lower limbs; however, he was generally well between the episodes. The possibility of a hereditary periodic fever syndrome was raised in 2004 and he underwent genetic screening for FMF and HIDS, which were again negative; no MEFV mutations were found by the National Amyloidosis Centre, and HIDS on the basis of normal levels of IgD and urinary mevalonic acid. The patient was treated empirically with prophylactic colchicine, and ibuprofen in the event of inflammatory episodes. He remained relatively well, but continued to experience episodic abdominal and musculoskeletal pains. It also transpired that the patient’s mother had been suffering from musculoskeletal pains and fevers, for which she had been treated with oral steroids. Consequently, further investigations (table 1) and screening for TRAPS was arranged for both patients.
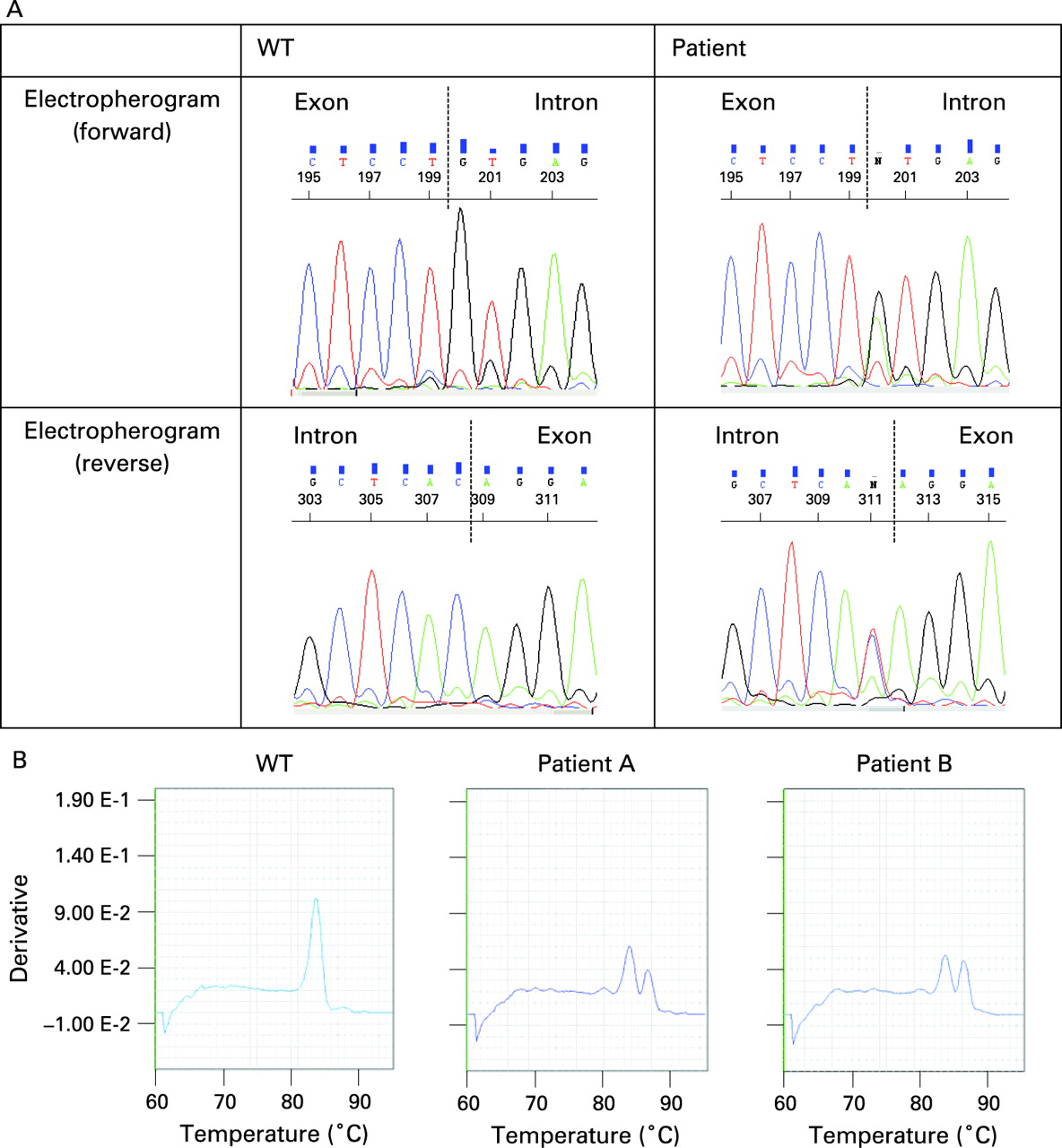
We found a novel mutation in the first base pair of intron 4 of TNFRSF1A, c.472+1G>A (C158delinsYERSSPEAKPSPHPRG); based on the crystallographic structure proposed by Banner et al,16 this would lead to a 45 nucleotide insertion of intronic DNA sequence into the mRNA, which in turn results in an in-frame insertion of 15 amino acids in the mature TNFR1 protein and deletion of a cysteine residue: C129 (158) in cysteine rich domain (CRD)3 of the mature TNFR1 protein (fig 1A). This mutation was not observed in 100 Caucasian controls and results in a clinical presentation of TRAPS in the proband and his mother. The effect of this mutation on transcription was studied by quantitative real-time PCR; wild type (WT) cDNA yielded a product with a dissociation temperature of 83.6°C (fig 1B); the proband’s cDNA, however, yielded a dissociation curve displaying two products, with melting temperatures of 83.6°C and 86.4°C respectively (fig 1B), indicating that a larger product was present in addition to the WT as a result of aberrant splicing. The presence of the larger product, approximately 130 base pairs (bp) in size, was confirmed on a 2% TAE agarose gel, in addition to the single 87 bp product of the healthy control (data not shown). To determine the exact nature of this insertion, primers TNFR 3–6 F and R were used to amplify exons 3 to 6 of the cDNA and the PCR products were sequenced. This showed two distinguishable products, identifiable as the WT allele and an allele with a 45-bp insertion from the fourth intron. This was also confirmed by subcloning of the two separate products (see Supplementary material). The effect of this insertion on the 3D protein structure and folding was examined using the modelling program, Modeller 9 V.1 (see Supplementary material).17 18 Comparative analysis of the 3D structure of the mutated protein (green) with wild type TNFR1 (blue), revealed the loss of the disulfide bond between C117 (146) and C129 (158) (red) and the introduction of an additional loop (pink).
Serum cytokine levels in patients with splice site mutation
Sera cytokine profiles in the patients with splice mutation were performed and compared with controls. The proband had significantly elevated IL1β, IL2, IL6, IL10 and IL12 levels, compared to healthy controls (n = 8) (table 2). TNF was marginally elevated but within the range of controls. In the proband’s mother, who was receiving treatment with steroids at the time, the levels of TNF, IL10 and IL12 were within the normal range, and only IL1β, IL6 and IL2 levels were elevated compared to controls (table 2).
NF-κB activation in PBMCs from patients with splice site mutation
We then investigated the activation of the NF-κB transcription factor in PBMC from patients with the splice mutation and compared the activation with PBMC from patients with TRAPS carrying the T50M (T79M) and C88R (C117R) mutations, respectively, and also patients with RA (fig 2). Basal nuclear expression of p50 and p65 NF-κB subunits were increased more than threefold in the patients with splice mutation compared with controls (n = 8). The nuclear expression of the p65 subunit in the splice mutations was comparable with that observed in the T50M (T79M) and C88R (C117R) mutations, yet less than that observed for patients with RA (n = 3). The p50 subunit nuclear expression was strongly elevated in the splice mutations compared with either controls or the T50M (T79M) and C88R (C117R) mutations and even greater than that observed for RA.

To determine how responsive the PBMC were to TNF stimulation, the nuclear expression of the p50 and p65 NF-κB subunits was measured following 5, 10 and 15 min of stimulation with 100 ng/ml TNF (fig 2). PBMC isolated from patients with splice mutation revealed a time-dependent increase in nuclear translocation of the p50 and p65 NF-κB subunits following TNF stimulation greater than with the controls. However, the increase in the p65 subunit nuclear expression in the splice mutations was less than that observed for patients with RA (fig 2). Conversely, the p50 nuclear expression was marginally greater in the splice mutation PBMC compared with patients with RA (fig 2). Interestingly, despite having elevated basal p65 nuclear expression in the PBMC carrying the T50M (T79M) and C88R (C117R) mutation, there was no further nuclear increase in p65 or p50 NF-κB subunits following stimulation with TNF in these mutations.
Soluble and cellular TNFR1 protein expression in patients with TRAPS with splice site mutation
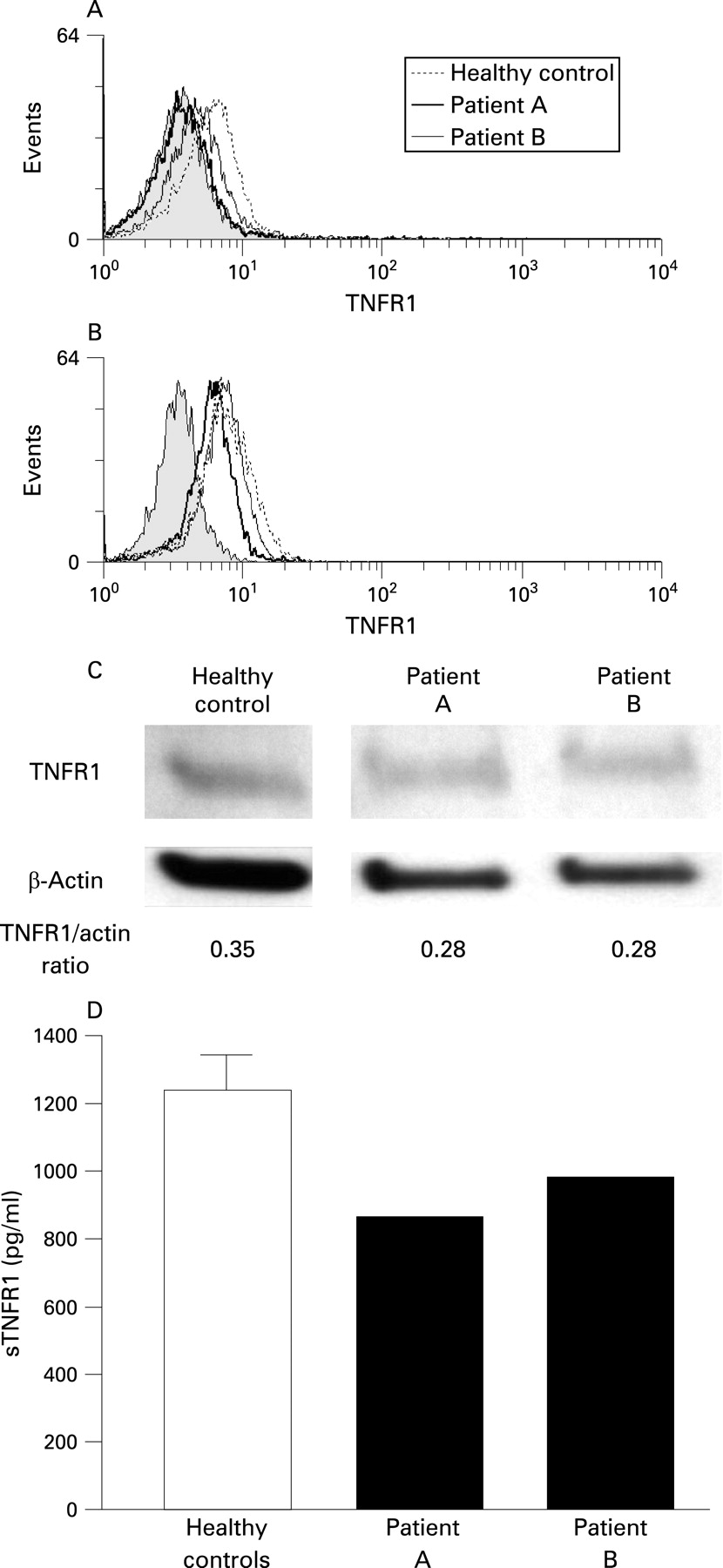
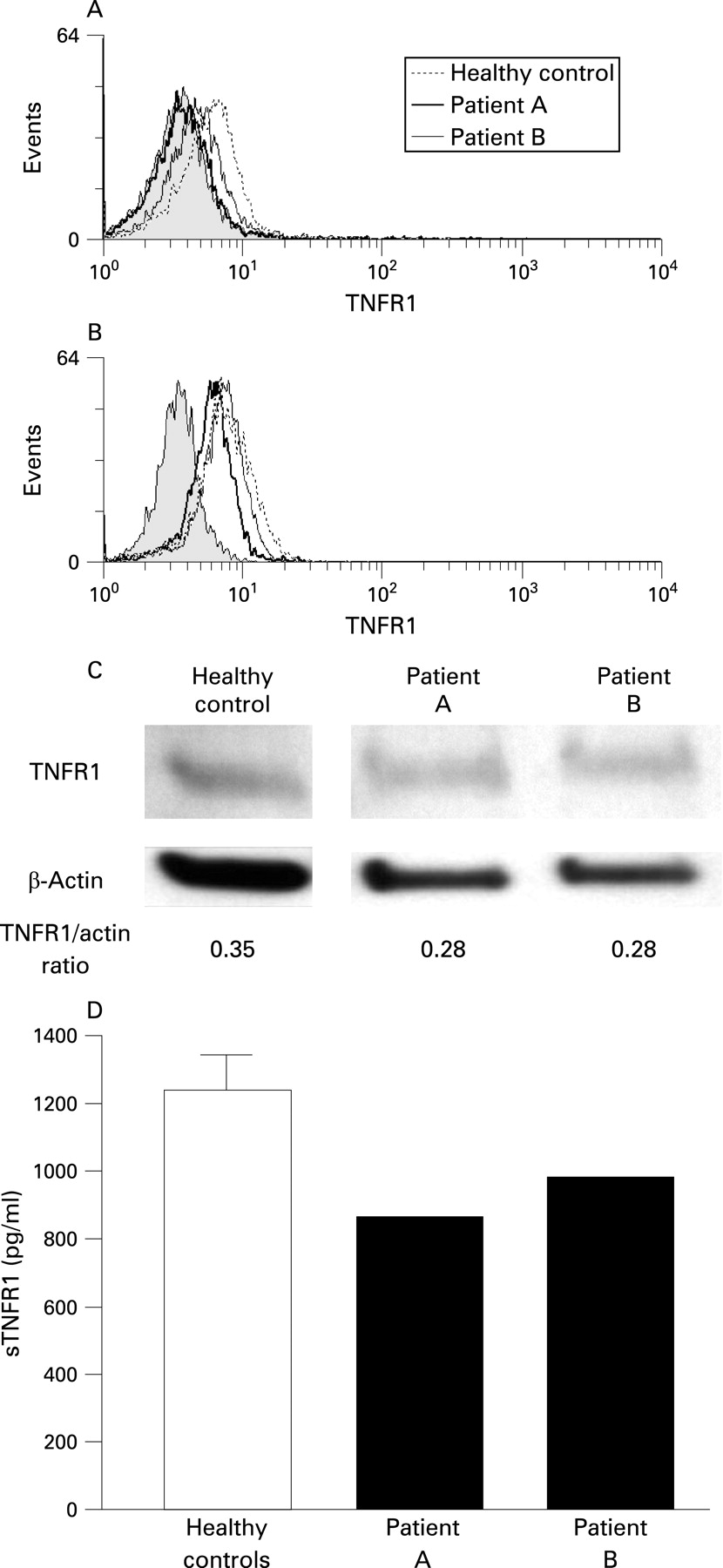
Given the multiple mechanisms proposed by which TNFSFR1A mutations can lead to clinical disease including impaired receptor shedding and dysfunctional trafficking, TNFR1 localisation was examined in the patients with splice mutation. Flow cytometric FACS analysis of the surface expression of TNFR1 on monocytes identified a reduction in the cell surface expression of TNFR1 on monocytes of the proband, patient A, (mean fluorescence intensity (MFI) = 3.64) and his mother, patient B, (MFI = 4.43) compared with the healthy controls (mean (SD) MFI = 5.20 (0.61), n = 5) (fig 3A). To determine whether the splice mutation induced the abnormal TNFR1 localisation that may underlie the reduced surface expression and elevated NF-κB activity, whole cell TNFR1 protein expression levels were analysed by intracellular staining (fig 3B). The reduction in TNFR1 expression was still evident in the patients with splice mutation (patient A, MFI = 6.19; patient B, MFI = 7.11) compared with healthy controls (mean (SD) MFI = 7.92 (0.38), n = 5).
{kind=link}
{kind=link}
{kind=link}
The reduction in TNFR1 protein expression was confirmed by WB analysis of lysates from PBMC. Both patients with splice mutation had marginally lower detectable TNFR1 compared with controls (fig 3C); densitometric analysis of the TNFR1 and β-actin (loading control) protein bands confirmed this.
This reduction in cell surface expression of TNFR1 and total protein was also reflected in the level of soluble TNFR1 (sTNFR1) in serum. Serum levels of sTNFR1 in the patients with splice mutation were decreased by almost a third in both patients compared with serum from controls (mean (SD) 1238 (234.5) pg/ml) (fig 3D). Analysis of TNFR1 shedding, assessed by FACS following stimulation of monocytes with phorbal 12-myristate 13-acetate (PMA) did not reveal a shedding defect (data not shown).
DISCUSSION
The majority of TNFRSF1A gene mutations identified underlying TRAPS are caused by missense mutations affecting cysteine residues involved in disulfide bonds and other residues in CRD1 and two that are predicted to have a pronounced affect on the overall secondary protein structure of TNFR1. In the present work, we have identified a novel TNFRSF1A mutation that is different to most TRAPS mutations as it introduces an insertion of 15 amino acids and the deletion of a crucial cysteine residue C129 (158) in CRD3 of TNFR1 protein. The only other known mutation in CRD3 causing TRAPS is an F112I mutation, found in affected members of a Finnish family.19 The mutation present in our patients at the 5′ end of intron 4 (c.472+1G>A) occurs at a critical splice junction recognition site, causing the nascent mRNA to be spliced incorrectly and results in the inclusion a portion of intron 4 in the mRNA. In the genomic DNA, a “gt” occurs 45 bases distal to the mutation in the intron sequence, thereby presenting a potential alternative splice site. The use of this site was confirmed by the increased product size observed on quantitative real-time PCR (gel and dissociation curve) and by DNA sequencing. The predicted protein, has an insertion of 15 amino acids, which would cause a size increase in the protein product from the WT 455 amino acids/50.5 kDa to 470 amino acids/52.2 kDa.
Based on the crystal structure of TNFR1 proposed by Banner et al16 this insertion at the splice site would cause an extended loop in the modelled 3D protein structure (see Supplementary material) and the deletion of a cysteine residue at position C129 (158), thus disrupting the disulfide bond between C117 (146) and C129 (158) in CRD3. Most of the previously described TRAPS-associated TNFRSF1A gene mutations occur within CRD1 and CRD2. CRD1, 2 and 3 share much homology, including six cysteine residues that form three disulfide bonds in each domain. The disulfide bonds formed by cysteine residues are crucial to the correct folding and secondary structure of TNFR1. Patients with TRAPS with greater clinical severity have been found to have mutations at key cysteine residues in CRD1 and CRD2 and it has been proposed that the substitution of cysteines for certain extracellular residues may lead to interchain disulfide-linked homodimerisation and constitutive activation of the receptor.14 15 20 Crystallographic studies of TNFR1 bound with lymphotoxin have predicted that CDR2 and CRD3 are largely responsible for mediating ligand binding.16 The deletion of C129 (158) disrupts the second disulfide bond in CRD3, which is homologous to the disulfide bond formed by C73:C88 (C102:C117) in CRD2. Mutations of these cysteines are known to have significant TRAPS pathology. The loss of C129 (158) in CRD3 by this splice site mutation may indeed cause constitutive activation in these patients, as reflected by their elevated serum cytokine levels and enhanced basal and TNF-induced NF-κB activation. The elevated level of IL1β observed in both patients (especially the proband), which would point to IL1 being a key mediator in the pathogenesis of TRAPS, and fits the observation that IL1ra treatment may be an alternative to anti-TNF therapy in TRAPS.21
Another significant observation was that TNFR1 cell surface expression, serum levels and cellular content, in the splice mutation, were all reduced. The reduced TNFR1 surface expression in these patients would argue against this mutation inducing a shedding defect, as found in some TNFRSF1A mutations including T50M (T79M) and C52F (C81F),1 3 and to a lesser extent H22Y (H51Y), C33Y (C62Y) and P46L (P75L).3 22 However, the degree of defective TNFR1 shedding in patients with TRAPS has been found to be quite variable between different mutations.5 Furthermore, no shedding defect has been observed in some patients with TRAPS in the presence of a reduced plasma concentration of sTNFR1, demonstrating further inconsistencies between TRAPS mutations. Analysis of a shedding defect in our patients did not reveal any significant defect (data not shown). The possibility that the reduced expression we observed might result from the loss of the antibody recognising epitope in the mutated TNFR1 receptor rather than loss of surface protein expression cannot be formally ruled out.
The overall reduced serum and TNFR1 cell surface expression observed in monocytes carrying the splice mutation is in keeping with several other TRAPS mutations, where similar findings have been reported.1 3 5 14 15 This supports the hypothesis that impaired intracellular trafficking of the misfolded mutated TNFR1 protein leads to its retention in the endoplasmic reticulum (ER) and ultimate disposal through a process of ER associated degradation (ERAD).14 15 Intriguingly, we did not observe any increase in cellular TNFR1 expression in patients carrying the splice mutation. Instead, total TNFR1 expression, as determined by intracellular staining and WB, was reduced in the patients with splice mutation compared with healthy controls, albeit to a lesser extent to that observed for surface expression. The possibility that differences observed between the patients with splice mutation and healthy controls could be due to loss of epitope binding must always be considered, but as a polyclonal TNFR1 antibody was used for WB, this would argue against this explanation. Although testing the ER retention hypothesis in primary cells is challenging, as there are currently no mutation-specific TNFR1 antibodies available to distinguish between WT and mutant TNFR1, future studies are planned using confocal microscopy to examine total TNFR1 intracellular localisation.
The mechanism whereby this splice mutation leads to the pro-inflammatory phenotype characteristic of TRAPS, is unclear. An important yet paradoxical feature of this splice mutation that despite having low cell surface TNFR1 expression, PBMC still retain the ability to respond to TNF compared with PBMC carrying the T50M (T79M) or C88R (C117R) mutation. The reasons why the splice mutation behaves so differently from T50M and C88R (T79M and C117R) in this regard are unclear. However, insertion of 15 amino acids at the splice site in these patients is likely to have significant structural implications for the receptor. Interestingly, there is increasing evidence that the p50 NF-κB homodimer can associate with the B cell lymphoma 3 (Bcl-3) transcriptional coactivator to confer transactivation capabilities23 thereby providing a potential mechanism by which increased activation of the p50 homodimer can be associated with promotion rather than inhibition of the pro-inflammatory responses and elevated serum cytokine levels we have observed.
In conclusion, much work over the past 5 years has been focused on investigating how mutations in the first two CRDs translate into receptor dysfunction. Identification of this novel splice site mutation affecting CRD3 suggests that these investigations should extend to include mutations in this domain. Considering the biochemical significance of the cysteine residues in all CRDs for proper structure and folding of this protein, in addition to the proposed role CRD3 plays in ligand binding, it is somewhat surprising that the patients carrying this mutation do not present with a more severe pathology. Further studies of this mutation and others are in progress to unravel the complex cellular mechanisms underlying the varying TRAPS pathologies and observed heterogeneity between different TNFRSF1A mutations.
Acknowledgments
We are particularly grateful to the patient and mother who agreed to participate in the study.
REFERENCES
Supplementary materials
web only appendix 67/11/1589
Files in this Data Supplement:
Footnotes
Funding: This study was supported in part by grants from the Wellcome Trust, the Sir Jules Thorn “Seed Corn” Fund, the Arthritis Research Campaign, the Charitable Foundation of the Leeds Teaching Hospitals and Bart’s and the London Charitable Foundation.
Competing interests: None declared.
Ethics approval: The study was approved by the Leeds (East) Research Ethics Committee.