
ABSTRACT
We present a case of a man with headache and progressive behavioural disturbance. His cognitive decline progressed over a few months such that he was unable to hold a conversation or carry out any daily tasks such as washing and dressing. He had some upper motor neurone signs in his limbs and features of brainstem dysfunction including dysarthria and ocular abnormalities. His brain magnetic resonance imaging showed signs of brain ‘sagging’. He was thought to have frontotemporal brain sagging syndrome. Prior to any treatment, he began to improve. Over the course of a week he became markedly better, was back to normal within 3 months and remains so 7 months later. We propose that resolution of spontaneous intracranial hypotension led to resolution of frontotemporal brain sagging syndrome. We believe this is the first case described where this has occurred without any intervention. It is important to recognise this condition as a potentially reversible cause of dementia.
Case presentation
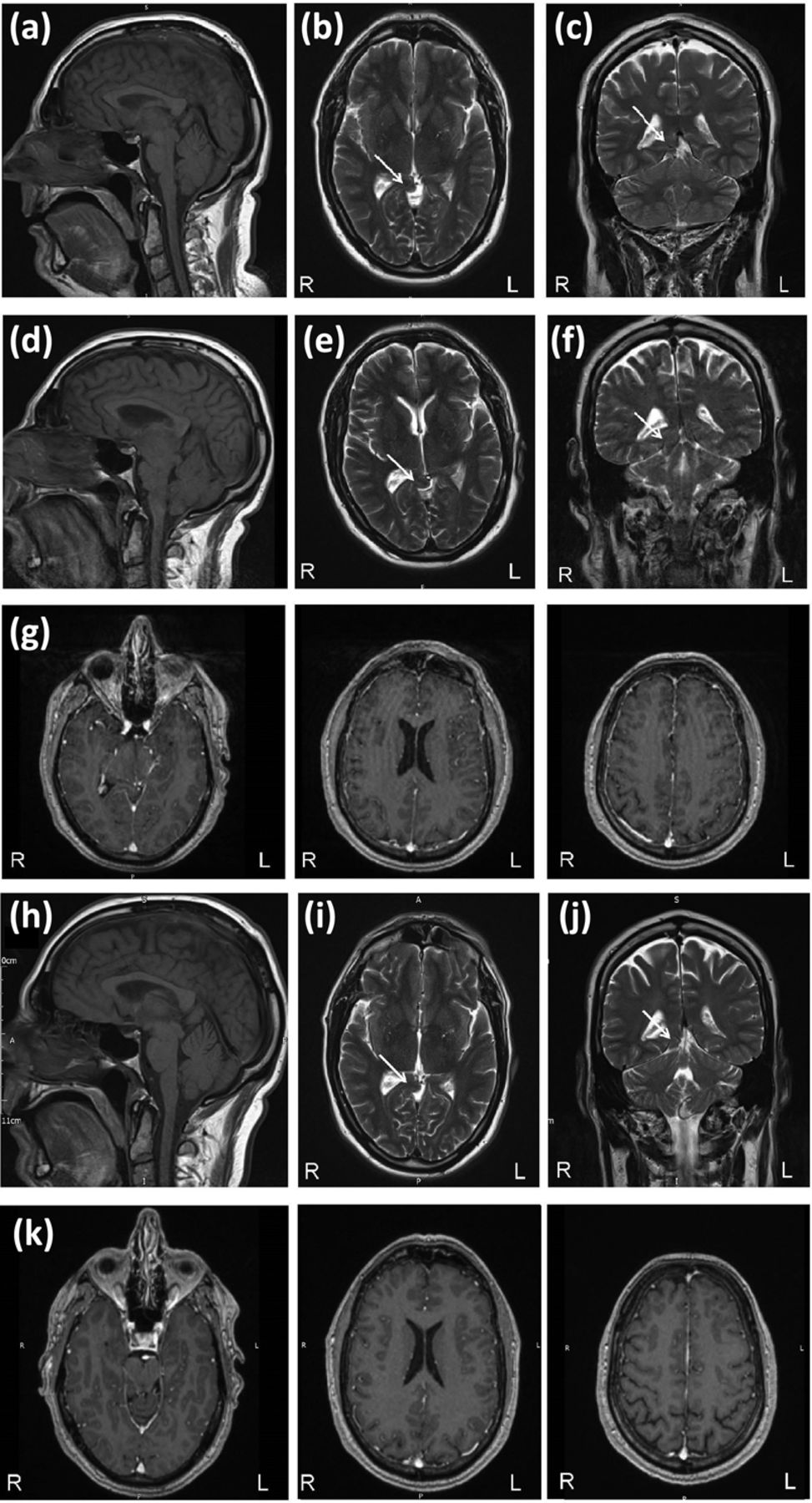
We report the case of a right-handed man who was initially seen aged 55. He had been well, lived with his family and owned a construction company. His first symptom was exertional headache, initially on hitting golf balls but then on a wider range of activities. Examination in June 2016 was normal but magnetic resonance imaging (MRI) showed transtentorial herniation of the medial temporal lobes (Figs 1a–c). This was thought to be longstanding and unlikely to be related. About a year later, his headaches became more constant, worse on standing and improved on lying flat. At about this time his family noted some behavioural changes. He initially had larger food portions but this progressed to cramming food in his mouth, sometimes having to spit it out, and abruptly leaving the table. He was over-familiar with people. He would wake at 2am and play the radio and television loudly, disturbing his family. He was restless, and increasingly unable to carry out his work effectively. He was admitted to hospital in January 2018 and found to be over-familiar, distractible and lacking insight. He had ocular flutter and extensor plantars but physical examination was otherwise normal. Blood test were normal, including HIV, syphilis, paraneoplastic antibodies (anti Hu, Yo, Ri) and genetics for Huntington's disease, MAPT, GRN, and C9orf72. Cerebrospinal fluid (CSF) showed an opening pressure of 10 cmH2O, white cell count 8 x 106/L, red cell count 2,380 x 106/L, glucose 4.4 mmol/L (no paired serum), protein 0.39 g/L. MRI showed some progression of the medial temporal herniation and meningeal enhancement (Figs 1d–g). Imaging of the spine did not reveal any site of a CSF leak. He scored 86/100 on the Addenbrooke's cognitive examination version 3 (ACE-III; attention 17/18, memory 19/26, fluency 9/14, language 26/26, visuospatial 15/16). During admission, his headache improved but he otherwise deteriorated. He developed dysphagia and dysarthria and became more apathetic. An examination 7 weeks after admission revealed hypomimia, jerky pursuit and hypometric saccades. He had a positive glabellar tap, jaw jerk and pout reflex, and a subtle grasp reflex on the right. His tongue was normal. He had stimulus-sensitive myoclonus to touch but not sound. There was no wasting or fasciculations. Upper limb examination revealed normal power but reflexes were brisk with positive finger jerks bilaterally, and he had bilateral dysdiadochokinesia. Lower limbs had increased tone bilaterally with normal power, brisk knee jerks and upgoing plantars bilaterally. He had normal Luria and applause test responses, and normal cognitive estimates. He could not attempt an ACE-III examination. At this time, his prognosis looked bleak. He was apathetic and was not able to care for himself. A further lumbar puncture 6 weeks after the first demonstrated CSF pressure of 7 cmH2O and normal constituents. He fulfilled the behavioural criteria for behavioural variant of frontotemporal dementia (bvFTD). He was discharged to a nursing home.

Magnetic resonance imaging brain appearances consistent with brain sagging. (a) T1 sagittal, (b) T2 axial and (c) T2 coronal from July 2016 showing transtentorial herniation of the medial temporal lobes. (d) T1 sagittal, (e) T2 axial and (f) T2 coronal from January 2018 showing some progression of the medial temporal lobe herniation, with (g) gadolinium enhancement of the meninges. (h) T1 sagittal, (i) T2 axial and (j) T2 coronal from August 2018 showing stable appearances of the temporal lobe herniation, and (k) resolution of the meningeal gadolinium enhancement. L = left; R = right; the arrow indicates the transtentorial herniation.
One week after the second LP, he started to engage more in conversation and activities. This improved over the following week, during which time he had a meeting with the nursing home manager to discuss his discharge. He describes this experience like ‘waking up’. At the end of that week, he was discharged home, and continued to improve. Three months later he was back to normal; free of headache, going to work, able to travel to London on the train and underground, and taking part in normal family life. He recalled very little about the period during which he was unwell. His neurological examination was normal and he scored 96/100 on the ACE-III (attention 18/18, memory 24/26, fluency 13/14, language 25/26, visuospatial 16/16). An MRI in August 2018 showed an improvement in the sagging appearance of the brainstem and resolution of the meningeal enhancement, but with stable medial temporal herniation (Figs 1h–k). Seven months after the start of his improvement, he remains well.
We propose that this patient had frontotemporal brain sagging syndrome (FTBSS). Wicklund et al described eight cases in 2011, characterised by progressive behavioural and cognitive decline, with a sagging appearance of the brain on MRI.1 Other notable features included headache, daytime somnolence, dysarthria and gait disturbance. As with our patient, four patients had transtentorial herniation of the medial temporal lobe structures, and 3/8 had meningeal enhancement. There was some response to treatments aimed at reversing the presumed low-pressure state, including steroids and blood patching, but this was not sustained in any patients. Schievink et al more recently described 29 cases with a long follow-up (average 7.85 years).2 Interestingly, 21/29 patients had a good long-term outcome, though a high proportion (18/29) underwent multiple surgical procedures. Also of note, recovery of this syndrome has been reported after head injury, where the authors propose that the patient's fall created a contusion and auto-blood patch.3
It is thought that intracranial hypotension causes the frontal syndrome. Symptoms of low-intracranial pressure tend to occur prior to those of behavioural disturbance, and features of brain sagging on MRI are universal. It appears that interventions aimed at resolving intracranial hypotension can improve cognitive and imaging abnormalities. This is perhaps most convincingly illustrated in the Schievink series where intrathecal saline infusion helped 7/8 patients.2 A proposed mechanism for this syndrome is that intracranial hypotension causes brain sagging, with distortion of the midline and brainstem anatomy. This is thought to disrupt the frontotemporal cortices or their networks, causing a clinical syndrome of bvFTD. Symptoms such as dysarthria and ocular abnormalities may reflect brainstem disturbance. Coma can also result, thought to be due to disruption of the brainstem reticular formation and/or the pontine tegmentum.4
Our patient had significant cognitive impairment and physical signs, which resolved without specific treatment. This may be due to spontaneous resolution of a CSF leak, or perhaps an ‘auto-blood patch’ from lumbar puncture. We note the high red cell count of 2,380 x106/L from the first lumbar puncture, which may have caused blood to infiltrate the CSF space.
This case of apparent ‘reversible dementia’ has been remarkable to witness. It is important to recognise FTBSS as a potentially treatable cause of cognitive decline.
- © Royal College of Physicians 2019. All rights reserved.








{kind=link}