


Abstract
Use of the popular club drug ecstasy (3,4-methylenedioxymethamphetamine, MDMA) can result in life-threatening hyperthermia and rhabdomyolysis. Recent studies show a link between skeletal muscle uncoupling proteins in MDMA-mediated hyperthermia. The mechanisms by which MDMA interacts with skeletal muscle mitochondria are largely unknown. The present study was designed to comprehensively evaluate the effects of MDMA on bioenergetics and toxicity of skeletal muscle. Using 31P nuclear magnetic resonance (NMR) and serum creatine kinase levels, we demonstrate evidence for uncoupling of oxidative phosphorylation in the skeletal muscle of MDMA (40 mg/kg)-treated rats. In vivo, rats treated with MDMA had significantly elevated serum creatine kinase levels, a marker of rhabdomyolysis, 4 h post-MDMA treatment (955 ± 132 IU/l) compared with saline-treated controls (373.2 ± 59 IU/l). β-ATP signal areas after MDMA treatment showed significant reductions (15%) from the baseline values with corresponding increases in inorganic phosphate (88% increases) and decreases in intracellular pH. Clark electrode experiments on isolated skeletal muscle mitochondria in vitro (1–5 mM MDMA) and ex vivo in MDMA-treated animals demonstrated no evidence of uncoupling of oxidative phosphorylation. In vitro experiments using L6 myotubules cocultured with primary hepatocytes demonstrated the presence of uncoupling protein-3 in the L6 myotubules, but no evidence of a direct effect of MDMA or its potential metabolites on cellular creatine kinase concentrations. These findings suggest that MDMA uncouples skeletal muscle mitochondria in vivo but that this uncoupling is the result of indirect mechanisms.
Abuse of the club drug 3,4-methylenedioxymethamphetamine (MDMA, Ecstasy) can result in life-threatening hyperthermia with maximum body temperature correlating with mortality (Gowing et al., 2002). Along with hyperthermia, persons with MDMA-induced toxicity can develop rhabdomyolysis, muscle rigidity (Dar and McBrien, 1996), renal failure (Fahal et al., 1992), and hyperkalemia (Ravina et al., 2004). Despite its popularity and prevalent usage, we have only recently begun to understand the thermogenic mechanisms of MDMA and how they contribute to human toxicities.
Hyperthermia induced by MDMA involves a complex interaction between the hypothalamic-pituitary-thyroid axis and the sympathetic nervous system (Sprague et al., 2003). Removal of either the pituitary or thyroid gland prevents MDMA-induced hyperthermia and paradoxically generates a hypothermic response (Sprague et al., 2003). Similarly, rats treated with MDMA have been shown to have elevated circulating T4 levels (Sprague et al., 2003), a finding that has been noted in human abusers of amphetamines (Morley and Shafer, 1982). MDMA-mediated thermogenesis is also dependent on activation of the sympathetic nervous system because antagonists of α1-adrenergic receptors (ARs) and β3ARs attenuate hyperthermia induced by MDMA when used alone and abolish it when coadministered (Sprague et al., 2003). Attenuation of hyperthermia by α1-antagonists such as prazosin may result from inhibition of MDMA-induced cutaneous vasoconstriction (Pedersen and Blessing, 2001). Alternatively, α1ARs potentiate the thermogenic effect of β3ARs in brown fat mitochondria (Zhao et al., 1997). This suggests that the action of α1ARs in skeletal muscle thermogenesis may also involve more than changes in regional blood flow (Zhao et al., 1997). Along with elevations in core temperature, rats treated with MDMA have significantly elevated temperatures in their skeletal muscle, which can be blocked by β3AR antagonists (Sprague et al., 2003). The thermogenic inhibitory properties of β3AR blockade in the skeletal muscle of MDMA-treated rats suggests a role for uncoupling proteins and nonshivering thermogenesis in MDMA-mediated hyperthermia.
Patients ingesting the herbicidal agents dinitrophenol (2,4-dinitrophenol) and pentachlorophenol have similar clinical presentations to MDMA toxicity. Symptoms that have been reported include rhabdomyolysis, muscle rigidity, hyperkalemia, metabolic acidosis, tachycardia, diaphoresis, and most commonly hyperpyrexia (Leftwich et al., 1982). The toxic molecular mechanisms of 2,4-dinitrophenol and pentachlorophenol have been well established; these protonophoric agents uncouple mitochondrial electron transport chain activity from ATP synthesis by inducing a leak of protons across the mitochondrial inner membrane into the mitochondrial matrix (Skulachev, 1998).
The primary site of nonshivering thermogenesis in rats is brown adipose tissue, not muscle, due to the activation of uncoupling protein 1 (UCP-1) (Nicholls and Locke, 1984). Although adult humans have little brown fat or UCP-1, UCP-3, a protein with significant homology to UCP-1, is abundantly expressed in human and rodent skeletal muscle (Boss et al., 1997). Like UCP-1, UCP-3 is regulated through the activation of peroxisome proliferator receptor elements and thyroid hormones (Gong et al., 1997; Teruel et al., 2000). Recently, UCP-3-deficient mice treated with MDMA were shown to have a blunted hyperthermic response and were resistant to its lethal effects (Mills et al., 2003). These findings strongly suggest that MDMA-mediated thermogenesis involves UCP-3 and the uncoupling of oxidative phosphorylation.
Although a link has been established between UCP-3- and MDMA-mediated hyperthermia, it is not clear whether MDMA is a direct uncoupler of mitochondrial respiration like 2,4-dinitrophenol, or whether its mechanisms are indirect. We recently demonstrated that the effects of MDMA on respiratory uncoupling in liver mitochondria are modest and only observed at concentrations far exceeding (>1000 times) that of serum levels in human users (Rusyniak et al., 2004). One possible explanation for this relative lack of effect may be that uncoupling proteins, although widely present in a variety of tissues, are not highly expressed in hepatocytes (Larrouy et al., 1997). The present study was designed to biochemically define the effects of MDMA on mitochondrial uncoupling in relation to MDMA-mediated hyperthermia, rhabdomyolysis, and ATP levels in skeletal muscle.
Materials and Methods
Materials. (±)MDMA was generously provided by the National Institute of Drug Abuse (NIDA) and was dissolved in water prior to usage. All other chemicals used were purchased from Sigma-Aldrich (St. Louis, MO).
Animals. Both naive and jugular vein cannulated (JVC), male Sprague-Dawley rats (175–300 g) were purchased from Harlan (Indianapolis, Indiana). All animals were housed in groups of two or three and given ad libitum access to food and drinking water. Housing conditions were maintained at a constant temperature of 22–24°C and a 12:12-h light/dark cycle. The JVC animals received complete cannula heparin (200 units/ml) maintenance upon arrival. Animals were acclimated for 1 week prior to exposure to MDMA. The care and handling of animals was conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The study was approved by the Institutional Animal Care and Use Committees of Indiana University School of Medicine and Ohio Northern University.
MDMA and Rhabdomyolysis in Rats. Jugular vein cannulated rats were divided into the following treatment groups: control (vehicle, n = 12) or MDMA (40 mg/kg s.c.; n = 10). Thirty minutes before and 4 h after MDMA administration, 500 μl of blood was collected per animal through the JVC cannula and replaced with an equal volume of saline. Each animal served as its own control for changes in baseline creatine kinase. Collected blood was allowed to clot for 30 min at room temperature and centrifuged at 10,000g for 10 min at 4°C. After serum was collected, the samples were immediately frozen at –80°C. Creatine kinase levels were determined by using the Vitros analyzer (Johnson and Johnson, New Brunswick, NJ), using Vitros creatine kinase slides. An 11-μl drop of sample was placed on the slide and distributed by the spreading layer to underlying layers. Reflection densities were monitored with a 670-nm wavelength during incubation (5 min at 37°C). The rate of change in reflection density was then converted to enzyme activity. The upper and lower detectable limits were 2000 and 20 IU/l, respectively.
In Vivo Measurement of ATP and Phosphocreatine in MDMA-Treated Rats. Rats were injected with urethane (1.75–2 g/kg s.c.), and once anesthetized, a 24-gauge angiocatheter was sutured into the peritoneal cavity and connected to a 60-inch long extension tube (total volume of 0.47 ml) allowing injections to be administered from outside the NMR spectrometer. Fiber optic temperature probes (FISO Commander, UMI version 1.9; FISO Technologies, Quebec, Canada) were placed 1 to 2 cm into the rectum and into the lateral hind leg of the limb without the NMR coil.
The animal was positioned on top of a water recirculating heating pad and gently secured inside a specially designed NMR probe constructed out of a 66-cm long section of 69-mm diameter acrylic tubing. A 0.9-cm diameter surface coil tunable to 106 MHz for 23Na and 161.8 MHz for 31P was placed directly over the lateral hind leg with a small glass bulb containing 400 mM phenylphosphonic acid (PPA) placed on top of the surface coil as an external reference for 31P spectra. Calculations of all phosphate peak areas were done with respect to PPA signal area.
All NMR experiments were performed on a 9.4 T, 89-mm vertical bore magnet interfaced to a Varian INOVA console (Varian, Inc., Palo Alto, CA). The magnet was shimmed using the 23Na signal to ∼60- to 80-Hz line width. Then, the surface coil was tuned to 161.8 MHz, and 31P spectra were collected using a 10-μs excitation pulse followed by acquisition of 3072 data points over a spectral width of 15 kHz. Two hundred fifty-six free induction decays were averaged over 4 min, 44 s for each spectrum. All NMR data were transferred to a personal computer and processed with NMR Utility Transform Software (NUTS; Acorn NMR Inc., Livermore, CA) for Windows 95/NT. 31P free induction decays were baseline corrected, zero-filling to 8 K data points, multiplied by a single exponential corresponding to 25 Hz line broadening, and Fourier transformed. Phosphocreatine and β-ATP resonance areas were measured by spectral integration between user-defined points.
Baseline muscle and core body temperatures along with steady-state phosphocreatine, β-ATP, and inorganic phosphate (Pi) signal areas were monitored every 5 min for 30 min after which MDMA (40 mg/kg in 0.2 ml of saline) was injected via the intraperitoneal catheter. Likewise, intracellular pH was measured based on the chemical shift of Pi relative to phosphocreatine as previously reported (Makos et al., 1998). Phosphorous data, pH, and temperatures were recorded every 5 min for the next 3 h. Each animal served as its own control for changes in temperatures and cellular phosphate levels and pH. Control values represented the mean of the measurements for 30 min prior to MDMA injection and were compared with the mean for each subsequent 30-min period for 3 h postinjection. ATP measurements were calculated only from the β-ATP peak as the other two nucleotide phosphate peaks contain contributions from ATP, ADP, and NAD. To be assured that our findings were not because of keeping the anesthetized animal in the magnet for the 210-min time period or other experimental conditions, three additional rats were examined under the same conditions with saline as a control.
Effects of MDMA on Mitochondrial Oxidative Phosphorylation. Mitochondria were isolated from the hind legs of adult male Sprague-Dawley rats using a method of protease digestion and differential centrifugation based on a previously reported protocol (Kerner et al., 2001). Final mitochondrial protein concentrations were determined by Bradford (1976) assay using bovine serum albumin as a standard.
Oxygen consumption in isolated mitochondria was monitored using a Clark oxygen electrode (Hansatech, Norfolk, England) kept at a constant 25°C with a computerized oxygraph. Reactions were performed in a reaction medium containing 125 mM potassium chloride, 4 mM potassium phosphate, 10 mM MOPS, and 2 mM magnesium chloride at a pH of 7.4. Experiments were conducted as described in Fig. 1 with the concentrations of MDMA being 0.1, 0.5, 1.0, and 5.0 mM for individual experiments. Rates of respiration as well as respiratory control index (RCI) and ADP/oxygen ratios were calculated according to standard procedures (Estabrook, 1967).

Experimental design: MDMA's effect on skeletal muscle mitochondria. The effects of MDMA were studied by calculating estimates of respiratory control and coupling efficiency in the absence and then presence of various concentrations of MDMA (0.1, 0.5, 1.0, and 5.0 mM) in reaction media containing either glutamate (5 mM) and malate (5 mM) or succinate (10 mM) and rotenone (2 μg). The representative graph (glutamate and malate) displays the experimental design with the additions being 0.5 mg/ml mitochondria (mito), 250 nmol of ADP, 0.1 mM MDMA, 250 nmol of ADP, and 40 μM dinitrophenol. Numbers indicate the individual rates of respiration (nanomoles of O2 per minute × milligrams of protein). State 2 was defined as the rate of respiration after the addition of substrates and mitochondria, state 3 as the initial burst of respiration after the addition of ADP, and state 4 as the return to baseline respiration after all of the ADP in state 3 has been converted to ATP. Each experiment was run in triplicate from three to four separate mitochondrial preparations. The effect of MDMA was assessed by comparing the respiratory rates for state 3, state 4, uncoupled state, and the RCI (state 3/state 4) and ADP/O (concentration of added ADP/oxygen consumed during state 3 respiration) ratios in the presence and absence of MDMA.
To determine the effect of MDMA on mitochondrial oxidative phosphorylation ex vivo, male Sprague-Dawley rats were allocated to receive either MDMA (40 mg/kg in 1 ml of H2O s.c.) or an equal volume of H2O. Animals were euthanized by decapitation 1 h after MDMA treatment with skeletal muscle mitochondrial isolation performed as described above. The experimental design is shown in Fig. 2.

Experimental design: ex vivo mitochondrial effects in MDMA-treated rats. Rats were treated with either MDMA (40 mg/kg s.c.) as in the above figure or an equal volume of saline. After 1 h, animals were sacrificed and mitochondria isolated. The representative graph (MDMA-treated animal) displays the experimental design with the additions being 5 mM glutamate (glu) and 5 mM malate (mal), 0.5 mg/ml mitochondria (mito), 250 nmol of ADP, 10 mM succinate (succ) and 2 μg of rotenone (rot), 250 nmol of ADP, and 40 μM dinitrophenol. Numbers indicate the individual rates of respiration (nanomoles of O2 per minute × milligrams of protein). Each experiment was run in triplicate from mitochondrial preparations obtained from six separate animals. The effect of MDMA was assessed by comparing the respiratory rates for state 3 and 4 along with the RCI and ADP/O ratios in animals treated with and without MDMA.
MDMA Biotransformation and Toxicity in Muscle Cell Culture. To determine the effects of MDMA metabolites on the toxicity of skeletal muscle cells, experiments were done with L6 cells with and without primary hepatocyte coculture. L6 cells were plated on 6-well tissue culture dishes in DMEM/Ham's F-12 complete media plus 10% FBS (DMEM/Ham's F-12 media containing 1.2 g/l sodium bicarbonate and supplemented with 200 units of penicillin, 200 μgof streptomycin, 2 μM dexamethasone, 1 μM insulin, and 10% fetal bovine serum). The cells were allowed to grow to >85 to 90% confluence at 37°C, 5% CO2, 95% O2. Myotubes were induced by replacing the media with DMEM/Ham's F-12 complete media plus 5% FBS. The change in media enhances myotube formation by increasing the fusion of myoblasts. The cells were allowed to grow in this myotube media for an additional 5 to 7 days before use, with myotube formation confirmed by microscopy.
Primary hepatocytes were isolated from male Sprague-Dawley rats using collagenase perfusion (Seglen, 1993) and plated at 4 × 105 cells per 0.4-μm inserts (BD Falcon, catalog no. 353493; BD Biosciences Discovery Labware, Bedford, MA) and placed in 6-well culture dishes containing DMEM/Ham's F-12 complete media plus 5% FBS (see above for contents) and incubated at 37°C, 5% CO2, 95% 02 with gentle rocking at 30 and 60 min postplating. Cell viability was determined via trypan blue exclusion and was always greater than 80%. The media was changed after 4 h, and the cells were then incubated overnight and used the next day.
Creatine kinase activity of the cells was determined using a modification of a previously described microtiter plate reader technique (Maglara et al., 2003). The myotubes were washed twice in 1 ml of Dulbecco's phosphate-buffered saline (DPBS) for 30 min each at 37°C, then harvested in 1 ml of DPBS with a rubber policeman and sonicated. The creatine kinase assay was carried out by mixing 50 μl of the sonicated cell sample and 200 μl of creatine kinase assay reagent (50 mM triethanolamine buffer pH 7.2, 2.5 mM glucose, 5 mM ADP, 10 mM AMP, 1 mM NADP, 1.5 kU/l glucose 6-phosphate dehydrogenase, 8 kU/l hexokinase, 20 mM phosphocreatine, 5 mM MgCl, and 1 mM dithiothreitol) in a 96-well plate. The absorbance of each well was measured at the 5- and 10-min wavelength of 340 nm using a microplate reader (Tecan Rainbow Thermo plate reader using the Tecan WinSeLecT microplate reader control and data analysis software). The 5-min time point was chosen as the first reading because preliminary work showed the initial reaction to have achieved linearity at this time point (data not shown). The activity of creatine kinase was calculated by spectrophotometrically measuring the production of NADPH from NADP+, where the rate of conversion of NADPH is directly proportional to creatine kinase activity (Maglara et al., 2003).
On the day of the experiment, L6 myotubes were washed twice in 1 ml of DPBS and tested alone or with hepatocyte coculture. For those experiments involving L6 myotubes alone, L6 myotubes were washed twice in 1 ml of DPBS and given either DPBS alone (control), 10 mM 2,4-dinitrophenol (positive control), 0.1. 0.5, 1, or 5 mM MDMA. 2,4-Dinitrophenol (10 mM) was used as a positive control, as it has been shown in a previous study to consistently reduce intracellular creatine kinase concentrations (Maglara et al., 2003). Cells were incubated for 3 h at 37°C, 5% CO2, 95% O2. For coculture experiments, L6 myotubes were given 1 ml of DPBS, and the inserts with hepatocytes were placed in the wells above them. The L6 hepatocyte cultures were then dosed with either DPBS alone (control), 10 mM 2,4-dinitrophenol (positive control), 0.1, 0.5, 1, or 5 mM MDMA. Cells were incubated for 3 h at 37°C, 5% CO2, 95% O2. After dosing, the inserts were discarded, and the myotubes were washed twice in 1 ml of DPBS for 30 min each time at 37°C. The cells were then harvested in 1 ml of DPBS with a rubber policeman and sonicated. Creatine kinase activity was determined as described above. To assure the viability of the primary hepatocytes in the experimental conditions, preliminary experiments were done incubating primary hepatocytes in DPBS for 3 h and measuring lactate dehydrogenase release and trypan blue exclusion. This data showed no evidence of increased lactate dehydrogenase release and greater than 75% cell viability (data not shown).
Western Blotting. L6 myotube protein (100 μg) were loaded into each lane of a BioRad minigel system. As a negative control, muscle mitochondria (75 μg of mitochondrial protein) from UCP-3–/– mice were used. As a positive control, recombinant murine UCP-3 (prepared in one of our laboratories) was used. The primary antibody was purchased from Chemicon International (UCP-3, AB-3046; Tecmecula, CA) and was incubated at a 1:1000 dilution; incubation was overnight at 4°C. The secondary antibody was a peroxidase-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and was incubated at a 1:500 dilution; incubation was for 1 h at room temperature. For detection, blots were processed using enhanced chemiluminescence kits (Amersham Biosciences Inc., Piscataway, NJ).
Statistics. A one-way analysis of variance model was used to assess the effect of MDMA concentration on the outcome measures in experiments with isolated mitochondria and L6 myotubes with a Dunnett's multiple comparison procedure to compare each of the MDMA concentrations to the control group. For ex vivo mitochondrial studies and serum creatine kinase experiments, serum creatine kinase concentrations, rates of respiration, and change in temperatures for animals treated with MDMA were compared using t test. Changes in phosphocreatine and β-ATP and temperatures in the NMR experiments were compared using repeated analysis of variance with each time point compared back to baseline using a Dunnett's multiple comparison procedure. Statistical significance was set a priori at a value of p ≤ 0.05.
Results
MDMA and Rhabdomyolysis in Rats. Rats treated with MDMA (40 mg/kg s.c.) developed stereotypical serotonin behavioral syndrome with significant increases in temperatures at 2 h from baseline in the MDMA-treated animals (37.2 ± 0.2°C to 39.1 ± 0.3°C; p = 0.0001). Serum creatine kinase levels, a marker of rhabdomyolysis, were significantly elevated 4 h post-MDMA treatment (955 ± 132 IU/l, p = 0.0002) compared with saline-treated controls (373.2 ± 59 IU/l).
In Vivo Measurement of ATP and Phosphocreatine in MDMA-Treated Rats. MDMA induced a rapid and significant rise in both core body and skeletal muscle temperatures. Core body temperatures increased from a baseline of 37.7 ± 0.14°C (mean ± S.E.M.) to 39.4 ± 0.29°C by 120 min with these temperatures sustained throughout the remaining 60 min (Fig. 3). Skeletal muscle temperatures similarly increased from a baseline of 37.1 ± 0.37°C to 38.9 ± 0.48°C by 150 min with elevated temperatures sustained the remainder of the experiment (Fig. 3). A representative spectra with integrations is shown in Fig. 4 with corresponding stacked bar graphs showing a steady decline in both β-ATP and phosphocreatine in a MDMA-treated rat compared with control. β-ATP signal areas showed significant (p < 0.05) reductions from the baseline values with the signal area decreasing 15% from baseline after MDMA treatment (Fig. 5). Decreases in β-ATP signal area were accompanied by corresponding increases in inorganic phosphate (88% increase) and decreases in intracellular pH (Fig. 5). The phosphocreatine signal also showed a small (8%) nonsignificant decrease after MDMA injection (Fig. 5). Three additional rats were examined to determine the effects of the experimental conditions on core and skeletal muscle temperatures and phosphocreatine and β-ATP signals using saline as a control. There were no significant changes in skeletal muscle and core body temperatures or phosphocreatine and ATP signal intensity over time (Figs. 3, 4, and 5).

MDMA's effects on core body (A) and skeletal muscle (B) temperatures measured during NMR experiments (Fig. 4). The values represent the mean ± S.E.M. for each 30-min period for control and MDMA-treated (40 mg/kg) rats. *, statistical difference compared with baseline (p < 0.05).

Representative NMR spectra and integrations with stacked plot graphs for phosphocreatine and β-ATP in an individual MDMA-treated and control rat. PPA served as an internal standard.

In vivo effects of MDMA on ATP, phosphocreatine, Pi, and intracellular pH. Changes in phosphocreatine (A), ATP (B), Pi(C), and intracellular pH (D) for MDMA-treated rats and controls. Values were obtained from the integration of 31P NMR spectra. A—C, each value represent the mean ± S.E.M. for each 30-min period divided by the mean of the 30-min baseline period. D, each value represents the mean intracellular pH ± S.E.M. for each 30-min period for both baseline and control. *, statistical difference compared with baseline values (p < 0.05).
Effects of MDMA on Mitochondrial Oxidative Phosphorylation in Isolated Mitochondria.Figure 1 illustrates the typical experiment testing the effects of increasing concentrations of MDMA on bioenergetic characteristics of isolated mitochondria from rat skeletal muscle. No differences were seen in the rates of respiration, RCIs, or ADP/O ratios between control and MDMA at concentrations from 0.1 to 1.0 mM for either complex 1 or complex 2 substrates (Table 1). At the highest MDMA concentration tested (5.0 mM), increases in state 4 respiration resulted in a significant decrease in the corresponding RCI (Table 1) compared with control. MDMA induced a slight decrease in the rate of 2,4-dinitrophenol (40 μM)-uncoupled succinate-fueled respiration (Table 1).
Effect of MDMA on mitochondrial respiration in isolated skeletal muscle mitochondria
The values represent the mean ± S.E.M. for three to four individual mitochondria preparations each run in triplicate. Values for state 3 and 4 are reported as nanomoles of O2 per minute per milligrams of protein.
Figure 2 illustrates the typical experimental results of MDMA's effect on mitochondrial respiration in the isolated skeletal muscle mitochondria of MDMA-treated rats. Rats given MDMA showed only a slight increase in complex 1-mediated RCI without significant differences seen in other parameters (Table 2).
Rates of oxidative phosphorylation in isolated skeletal muscle mitochondria of rats treated with MDMA
Mitochondrial respiration in animals treated with and without MDMA (40 mg/kg s.c.). The values represent the mean ± S.E.M. for six individual mitochondria preparations each run in triplicate. Values for state 3 and 4 are reported as nanomoles of O2 per minute per milligrams of protein.
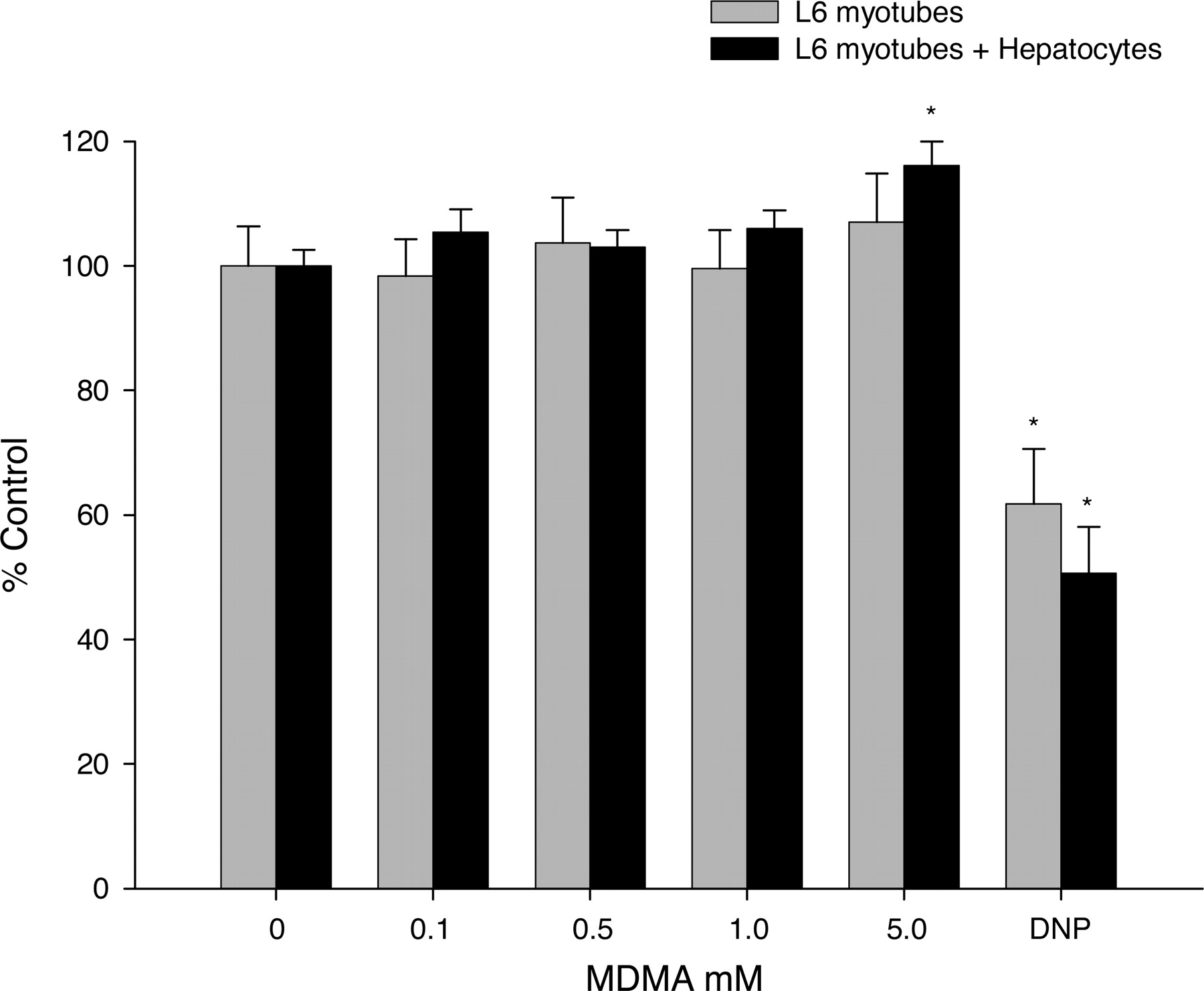
MDMA Biotransformation and Toxicity in Muscle Cell Culture. Increasing concentrations of MDMA did not show any reductions in intracellular creatine kinase in either L6 myotubes cultured alone or cocultured with primary hepatocytes, whereas 2,4-dinitrophenol significantly decreased intracellular creatine kinase content (Fig. 6).
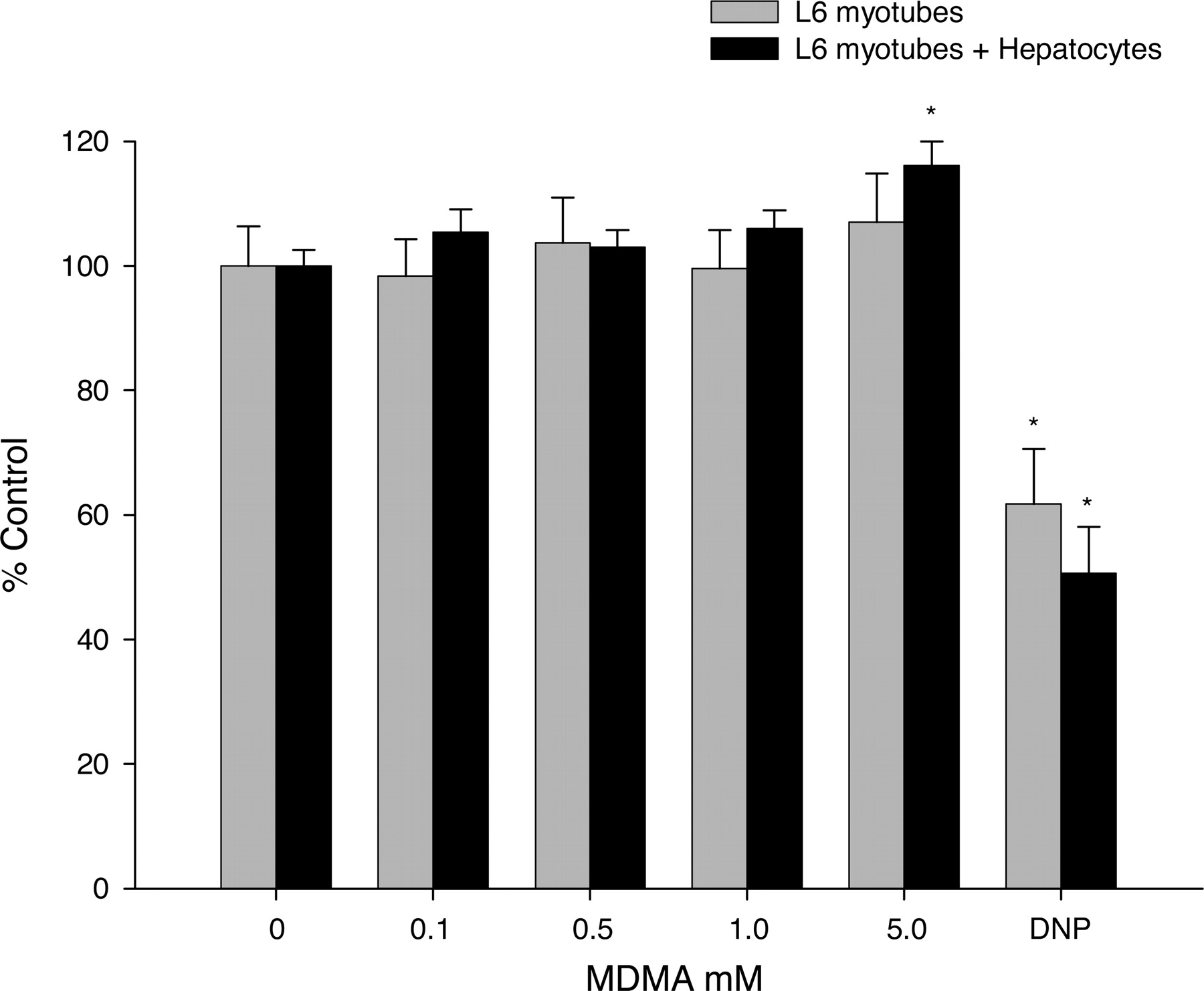
Effects of MDMA alone and with hepatocyte coculture on skeletal muscle cell culture creatine kinase activity. Bars represent the mean percent control creatine kinase activity (% control) ± S.E.M. for L6 myotubes alone (gray bars) or cocultured with primary hepatocytes (black bars). In both experiments, myotubes were exposed for 3 h to increasing concentrations of MDMA (0.1, 0.5, 1.0, and 5.0 mM) or dinitrophenol (10 mM) as a positive control. *, statistical difference compared with control (p < 0.05).
UCP-3 Protein Expression in L6 Myotubes. The results shown in Fig. 7 demonstrate that UCP-3 protein is expressed in L6 myotubes 5 days after the induction of differentiation.

Western blot of UCP-3 in L6 myotubes. Lane 1, 100 μg of cellular protein from L6 myotubes; lane 2, 75 μg of mitochondrial protein from muscle of UCP-3(–/–) mice; lane 3, recombinant UCP-3. Mitochondrial protein from UCP-3(–/–) mice served as a negative control; recombinant mouse UCP-3 served as a positive control. The latter migrates at 39 kDa, rather than 34 kDa, due to a 5-kDa fusion peptide.
Discussion
Agents which uncouple oxidative phosphorylation can result in severe and life-threatening hyperthermia. Recent studies have established a link between MDMA-induced hyperthermia, UCP-3, and β3ARs (Mills et al., 2003; Sprague et al., 2003). Although a role for mitochondrial uncoupling has been suggested to play a role in the hyperthermia induced by MDMA, to date there have been no studies, which have defined biochemically, the action of MDMA on mitochondrial uncoupling. In a previous study, we demonstrated that MDMA has very little effect on respiratory coupling in isolated liver mitochondria (Rusyniak et al., 2004). Conclusions based upon these findings are limited, however, because UCPs are not abundant in hepatocytes (Larrouy et al., 1997). Here we focused on the effects of skeletal muscle mitochondria, where UCP-3 is predominantly expressed (Boss et al., 1997).
Myocytes rely on oxidative phosphorylation and the hydrolysis of high-energy phosphate bonds from phosphocreatine in the formation of ATP. Alterations in the concentration of either phosphocreatine or ATP can result in energy requirements exceeding ATP production ultimately leading to muscle cell breakdown and rhabdomyolysis (Curry et al., 1989). Rhabdomyolysis is one of the most commonly reported complications in human users of MDMA (Dar and McBrien, 1996). Here, we show similar effects in rodents treated with MDMA. Four hours after MDMA treatment, serum creatine kinase levels were nearly 3-fold higher in the MDMA-treated animals. Other amphetamines, including methamphetamine and amphetamine, also cause rhabdomyolysis in human users (Kendrick et al., 1977; Richards et al., 1999). Consistent with ATP depletion, muscle rigor and hyperkalemia are also reported as part of the panoply of symptoms mediated by MDMA toxicity (Dar and McBrien, 1996; Ravina et al., 2004). In our experiments, we measured total creatine kinase activity and did not differentiate the various isoforms. It is possible that other creatine kinase isoforms, including MB, may have also contributed to the total creatine kinase increases we saw. A recent study showing structural damage to the skeletal muscle of MDMA-treated mice, however, supports that the creatine kinases in our study are secondary to skeletal muscle release (Duarte et al., 2005).
Previous investigators have shown rapid and transient decreases in ATP in the mouse striatum after multiple doses of methamphetamine, a phenylethylamine similar to MDMA (Chan et al., 1994). Methamphetamine in a cumulative dose of 40 mg/kg over 6 h, resulted in a 20% reduction in ATP concentrations (Chan et al., 1994). Along with decreases in ATP, methamphetamine administration increases concentrations of extracellular lactate (Stephans et al., 1998), glucose, and brain glycogen (Darvesh et al., 2002) suggesting an acute and rapid derangement in mitochondrial energy production. Similar effects on glycogen content have also been noted in studies with d-amphetamine and other amphetamine analogs (Nahorski and Rogers, 1973; Nowak, 1988). Although these prior studies implicate an effect of stimulants on energy production in the brain, recent work has also shown a role for skeletal muscle in MDMA-mediated hyperthermia (Mills et al., 2003). Using 31P NMR, a well recognized noninvasive tool for measurement of bioenergetic processes, we observed a 15% reduction in the skeletal muscle ATP signals 3 h after a single 40 mg/kg MDMA dose. These decreases were seen in conjunction with an 88% increase in inorganic phosphate and a reduction in intracellular pH. Although to a lesser extent (8%), the signal for phosphocreatine also showed a similar trend toward reduction after MDMA treatment. These data, when viewed in light of the corresponding skeletal muscle hyperthermia, strongly suggests impairment of mitochondrial oxidative phosphorylation. Because these animals were anesthetized and immobile, the effects of MDMA seen in this study are more consistent with a reduction in energy supply instead of excess consumption. In support of this, a study by Gordon et al. (1991) showed large increases in the metabolic rates of MDMA-treated (30 mg/kg) rats and a similar study a rapid increase in oxygen consumption paralleling the rise in core body temperature with 1 mg/kg methamphetamine (Makisumi et al., 1998). Likewise, our findings are similar to those obtained from rats treated with the classic uncoupler 2,4-dinitrophenol (Byrne et al., 1985; Jucker et al., 2000). We believe these prior findings along with our NMR data support a role for uncoupling of oxidative phosphorylation from MDMA. However, there are other possible explanations for our results, one being a decreased blood flow to the lower extremities. Although we did not measure blood flow, the skeletal muscle hyperthermia seen suggests well perfused tissues. Other researchers have shown increases in the mean arterial pressure in conscious animals suggesting increased perfusion (Badon et al., 2002). The animals in the present study were anesthetized and this may have affected perfusion. Urethane can lower blood pressure, but depends on the route of administration with these effects not seen with subcutaneous urethane as administered in our study. Because hypoxia and hypoperfusion can both decrease ATP and phosphocreatine concentrations, these possibilities should be explored in future experiments.
One of the most perplexing findings in the present study is the relationship between the ATP signal area and the relative lack of decrease seen in the phosphocreatine signal area. Because phosphocreatine and ATP are linked in the Lohmann or phosphocreatine reaction we would have expected to see far greater drops in the phosphocreatine signal area. One explanation for this is the possibility of a reversible inhibition of creatine kinase activity. Although we saw no evidence of this in our L6 myotube experiments (Fig. 6), it is possible that the effects were lost during the washing of the cells in preparation for measuring creatine kinase activity. Future studies will need to asses MDMA's effects, if any, on creatine kinase activity. Another explanation for the phosphocreatine and ATP discrepancy would be the disappearance of ATP from the metabolic pool, by the conversion to AMP, IMP, or cAMP. Although we have no data to suggest this, studies have shown that the combination of thyroid hormones and norepinephrine can cause significant increases in intracellular cAMP in brown adipocytes (de Jesus et al., 2001). Because MDMA has been shown to increase both T4 and norepinephrine it is conceivable their interactions with either UCP-1 or UCP-3 or both could account for some of the loss of ATP through its conversion to cAMP. Finally, although not significant, we do see decreases in phosphocreatine concentrations (8%) that parallel the time course of those seen in ATP. The statistical variability seen in the phosphocreatine measurements could easily account for some of the discrepancy seen.
Despite the findings above, we found little evidence of a direct effect of MDMA on the bioenergetics of isolated skeletal mitochondria. Although a small nonsignificant increase was noted for complex 1 and 2 substrates in state 4 respiration with a corresponding decrease in the RCI values, the magnitude of this effect was small when compared with 2,4-dinitrophenol. Only at MDMA concentrations corresponding to greater than 1000 times those found in human cases did we note any significant effects. These effects were noted only for the RCI and not state 4 respiration, which is the respiratory state most effected by uncoupling. The mitochondria of MDMA-treated animals likewise showed similar respiratory parameters to controls with the minimal amount of uncoupling seen in the in vitro mitochondrial isolations no longer evident in the treated animals. These differences may be due to the lower concentrations of MDMA that the skeletal muscle mitochondrial were likely exposed to in the treated animals compared with 5 mM in the in vitro studies. In isolated mitochondria work, however, results must be interpreted with some caution as the experimental conditions may result in required ions and/or cofactors being lost or altered during the preparation. The main reason for our testing MDMA in the isolated mitochondrial preparations was to elicit whether it had any direct uncoupling activity. Because amphetamines can cross membranes in both their neutral and protonated species by cycling across the membrane, they could act as a classic uncoupler. In high concentrations, this has been demonstrated with chlorphentermine, another phenylethylamine (Zychlinski and Montgomery, 1985). Given that we saw no significant evidence of uncoupling in our preparations at concentrations as high as 5 mM is indicative that this classic uncoupling is not likely responsible for the effects we saw in the in vivo studies. If MDMA works through βAR, as we hypothesize, we would not expect to see evidence of this in the isolated mitochondria which lack a cell membrane and hence βAR. Together, the data from these experiments suggest that direct effects of MDMA on mitochondria are minimal and are not likely responsible for the physiological responses seen in vivo.
MDMA and its metabolites, including methylenedioxyamphetamine, produce hyperthermia (O'Callaghan and Miller, 1994). To determine a possible direct effect of MDMA or its metabolites on the skeletal muscle, we employed a coculture assay with L6 myotubes and primary hepatocytes. In this study, we demonstrated the presence of UCP-3 by Western blot in L6 myotubes (Fig. 6). Previous studies have only demonstrated the presence of UCP-3 mRNA in L6 myotubes (Nagase et al., 1999). The L6 cell line has also been used as a model of myotoxity with similar findings in vitro to known in vivo effects of toxins (Kato et al., 1992). Because amphetamines (Green et al., 1986) and MDMA (Lim and Foltz, 1988) are extensively metabolized by the liver, we employed a model of coculture with primary hepatocytes and L6 myotubes. In our experiments, we saw no evidence of toxicity from either MDMA alone or in coculture despite the use of concentrations as high as 5 mM. This finding is in contrast to the results we obtained in rats treated with MDMA, in which serum creatine kinase increased nearly 3-fold and would suggest a more integrative mechanism of toxicity not replicated in our in vitro studies. However, 2,4-dinitrophenol produced marked toxicity as has been previously reported (Maglara et al., 2003). Although 2,4-dinitrophenol has no known interactions with uncoupling proteins, it uncouples oxidative phosphorylation with adverse effects in humans which parallel those of MDMA. It is important to mention, however, that since we did not measure oxidative phosphorylation directly in these cells it is possible that there was some mild uncoupling that was insufficient enough to affect creatine kinase release in our in vitro model. L6 cells also have significant differences with mature skeletal myofibers including slower differentiation, absence of insulin growth factors, and a more limited number of myogenic cell markers (Neville et al., 1997). We believe, however, that the demonstration of UCP-3 in this cell model and previous work showing that UCP-3 in L6 cells is regulated by thyroid hormones and βAR agonists (Nagase et al., 1999, 2001) similar to mature skeletal muscle makes this line a suitable model for in vitro work with MDMA.
The dependence on UCP-3 for skeletal muscle hyperthermia induced by MDMA (Mills et al., 2003) and the lack of a direct effect observed in the current study suggests that MDMA's effects on uncoupling proteins are indirect. Because skeletal muscle hyperthermia is attenuated by antagonists of β3 adrenergic receptors (Sprague et al., 2003), the β3AR agonist, norepinephrine, may mediate the mitochondrial effects seen with MDMA. Similar to UCP-1, UCP-3 is regulated by β3ARs through norepinephrine and thyroid hormones (Gong et al., 1997). Thyroid hormones work in concert with the sympathetic nervous system, amplifying intracellular levels of cyclic AMP from βAR stimulation (de Jesus et al., 2001). Together, the actions of the thyroid and sympathetic nervous system result in a synergistic, acute activation of uncoupling proteins, with corresponding increases in respiration up to 450% in brown adipose tissue and 35% in skeletal muscle (Zaninovich et al., 2003). This hypothesis is supported in human users of MDMA who have been shown to have higher levels of plasma norepinephrine and dopamine compared with nonusers (Stuerenburg et al., 2002). Similarly, rats treated with the same 40 mg/kg dose of MDMA used in this study, also demonstrate a 35-fold increase in plasma norepinephrine levels (Sprague et al., 2003). Along with activation of β3ARs, norepinephrine acts as an agonist at α1ARs augmenting brown fat-mediated thermogenesis. Agonists of α1ARs also mediate vasoconstriction of cutaneous blood vessels impairing heat dissipation through an effect known to contribute to the hyperthermia induced by MDMA (Pedersen and Blessing, 2001).
Whereas previous studies have demonstrated the importance of mitochondrial uncoupling proteins in MDMA-induced skeletal muscle hyperthermia, the mechanisms by which MDMA interacts with skeletal muscle mitochondria were heretofore unknown. We have shown that MDMA appears to uncouple oxidative phosphorylation in vivo, likely through an indirect mechanism independent of metabolism resulting in reductions in both ATP and phosphocreatine with subsequent development of hyperthermia and rhabdomyolysis.
Footnotes
-
This study was supported by a Clarian Value Funds grant, Clarian Health Systems, Indianapolis, Indiana.
-
Part of this work was presented at the 2004 Society for Academic Emergency Medicine Meeting, Orlando, Florida, May 2004.
-
doi:10.1124/jpet.104.079236.
-
ABBREVIATIONS: MDMA, 3,4-methylenedioxymethamphetamine; AR, adrenergic receptor; UCP, uncoupling protein; JVC, jugular vein cannulated; PPA, phenylphosphonic acid; MOPS, 4-morpholinepropanesulfonic acid; RCI, respiratory control index; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; DPBS, Dulbecco's phosphate-buffered saline; ADP/O, ADP to oxygen ratio.
- Received October 13, 2004.
- Accepted January 10, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}