


Abstract
Heterozygous null mutations in the melanocortin-4 receptor (MC4R) cause early-onset obesity in humans, indicating that metabolic homeostasis is sensitive to quantitative variation in MC4R function. Most of the obesity-causing MC4R mutations functionally characterized so far lead to intracellular retention of receptors by the cell's quality control system. Thus, recovering cell surface expression of mutant MC4Rs could have a beneficial therapeutic value. We tested a pharmacological chaperone approach to restore cell surface expression and function of 10 different mutant forms of human melanocortin-4 receptor found in obese patients. Five cell-permeant MC4R-selective ligands were tested and displayed pharmacological chaperone activities, restoring cell surface targeting and function of the receptors with distinct efficacy profiles for the different mutations. Such mutation-specific efficacies suggested a structure-activity relationship between compounds and mutant receptor conformations that may open a path toward personalized therapy. In addition, one of the five pharmacological chaperones restored function to most of the mutant receptors tested. Combined with its ability to reach the central nervous system and its selectivity for the MC4R, this pharmacological chaperone may represent a candidate for the development of a targeted therapy suitable for a large subset of patients with MC4R-deficient obesity.
Introduction
Disease-causing mutations in G protein-coupled receptors (GPCRs) often lead to decreased cell surface expression and concomitant loss of function as a result of improper folding (Schöneberg et al., 2004; Thompson et al., 2008). These mutant receptors, generally recognized by the cell's quality control system within the endoplasmic reticulum (ER) and Golgi apparatus, are retained intracellularly and targeted for degradation. In many of these conformational diseases, the mutation occurs in receptor domains that do not directly affect ligand binding or G protein coupling, opening the possibility for interventions that could restore receptor function by rescuing folding and cell surface expression (Bernier et al., 2004; Conn et al., 2007). Such functional rescue has been achieved for several GPCRs, indicating that pharmacologically selective compounds, termed pharmacological chaperones (PCs), can stabilize the misfolded receptors to facilitate their export from the ER to the plasma membrane where they are active (Morello et al., 2000; Petäjä-Repo et al., 2002; Noorwez et al., 2004; Bernier et al., 2006; Robben et al., 2007; Conn and Janovick, 2009). The clinical effectiveness of a PC approach has been tested for one such disease, nephrogenic diabetes insipidus, where a vasopressin antagonist, acting as PC, rescued the function of ER-retained V2-vasopressin receptor mutants and significantly improved the kidney function of patients with nephrogenic diabetes insipidus (Bernier et al., 2006).
PCs may present an attractive therapeutic option for severe early-onset morbid obesity that results from mutations in the melanocortin-4 receptor (MC4R), a receptor that plays a pivotal role in energy homeostasis (Cone, 2005). In humans, MC4R mutations lead to an obese phenotype similar to the homozygous null-mouse model (Huszar et al., 1997; Chen et al., 2000) and represent the most common monogenic cause of severe early-onset obesity (Farooqi and O'Rahilly, 2006). To date, ∼80 distinct mutations of MC4R have been reported in the obese human population. One MC4R antagonist was previously shown to have pharmacological chaperone action on two MC4R mutants (Fan and Tao, 2009). However, the large diversity of obesity-related trafficking-defective mutations in MC4R calls into question the ability of a single compound to restore cell surface expression and function to all mutant forms. This idea is reinforced by the fact that mutations, which are broadly distributed throughout the receptor structure, might lead to different conformational changes. To address this question and determine whether different PC candidates show different efficacies toward distinct mutants, we tested five cell-permeant MC4R antagonists that belong to three structurally distinct chemical classes. The compounds were studied for their ability to rescue cell surface expression and signaling activity of 10 naturally occurring mutant forms of MC4R that cause obesity (Tao, 2005; Tan et al., 2009). Clear differences were found in the efficacies and potencies between compounds on each mutant, revealing unique rescue profiles for individual PCs. It is noteworthy that one compound emerged as the most universal PC, rescuing nearly all mutations with the highest potency and efficacy. Furthermore, its bioavailability in brain, its clearance properties, and its receptor subtype selectivity make it a good candidate for the development of a clinically useful drug to treat genetic obesity caused by distinct MC4R mutations in humans.
Materials and Methods
Generation of Mutant Human Melanocortin-4 Receptor Constructs.
Ten mutant forms of human MC4R (hMC4R) [S58C, E61K, N62S, I69T, I125K, T162I, R165Q, R165W, C271Y, and P299H] were double-tagged with a 3×HA tag at the N terminus and a venus yellow fluorescent protein (vYFP) tag at the C terminus (see Supplemental Materials and Methods).
Compound Synthesis.
Compounds selected in the study were synthesized at Amicus Therapeutics (Cranbury, NJ) according to procedures described previously: 2-(2-(2-methoxy-5-nitrobenzylthio)phenyl)-1,4,5,6-tetrahydropyrimidine (MTHP) (Maguire et al., 2002); 3-(4-(2-(4-fluorophenyl)-2-(4-methylpiperazin-1-yl)ethyl)piperazin-1-yl)-2-methyl-1-phenylpropan-1-one (PPPone) (Arasasingham et al., 2003); 2-(5-bromo-2-methoxyphenethyl)-N-(N-((1-ethylpiperidin-4-yl)methyl)carbamimidoyl)-3-fluorobenzamide (MPCI) (Chaki et al., 2005); N-((2R)-3(2,4-dichlorophenyl)-1-(4-(2-((1-methoxypropan-2-ylamino)methyl)phenyl)piperazin-1-yl)-1-oxopropan-2-yl)propionamide (DCPMP) (Pontillo et al., 2005a); 1-(1-(4-fluorophenyl)-2-(4-(4-(naphthalene-1-yl)butyl)piperazin-1-yl)ethyl)-4-methylpiperazine (NBP) (Vos et al., 2006).
Structural Modeling.
The homology modeling of the inactive conformation of human MC4R was done as described previously (Tan et al., 2009) using β2-adrenergic receptor fused with T4 lysozyme (Protein Data Bank ID 2rh1) as a structural template. Three-dimensional structures of five small-molecule antagonists (including four stereoisomers of PPPone and two stereoisomers of both DCPMP and NBP) were generated by QUANTA (Accelrys, San Diego, CA). pKa values for charged groups were calculated by Marvin software (http://www.chemaxon.com/marvin/sketch/index.html). Conformational analysis of the ligands was performed using grid scan search for torsion angles of all rotatable bonds. The lowest-energy conformations of all stereoisomers of all ligands (except one stereoisomer of NBP with ΔE = 2.2 kcal/mol) were used for docking (see Supplemental Materials and Methods).
Transfection and Cell Culture.
HEK293T cells were transiently transfected with plasmids encoding chimeric WT or mutant hMC4Rs using FuGENE 6 (Roche Applied Science, Laval, QC, Canada) as the transfection agent. Cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, and 100 U/ml penicillin/streptomycin for 24 h. Transiently expressing cells were incubated in the presence or absence of antagonist for 12 h before flow cytometry or cAMP detection assays.
Quantitative Assessment of WT and Obesity-Associated Mutant hMC4R Membrane Expression by Flow Cytometry.
Cells were harvested 48 h after transfection, rinsed once in 1× Dulbecco's PBS (D-PBS), and transferred into 1× Tyrode's solution (140 mM NaCl, 2.7 mM KCl, 1 mM CaCl2, 12 mM NaHCO3, 5.6 mM d-Glucose, 0.49 mM MgCl2, 0.37 mM NaHPO4, and 25 mM HEPES, pH 7.4) supplemented with 1% BSA (Sigma-Aldrich, Oakville, ON, Canada) (Tyrode/BSA) containing mouse monoclonal anti-HA antibody (1:1000; HA.11; Covance, Burlington, ON, Canada) to label cell surface receptors. After 1-h incubation at room temperature, cells were washed once and resuspended in Tyrode/BSA containing anti-mouse Alexa Fluor 647 secondary antibody (1:1000; Invitrogen Canada Inc., Burlington, ON, Canada). After 1-h incubation at room temperature, cells were washed with Tyrode/BSA, resuspended in Tyrode's solution, and kept on ice. Just before analysis, propidium iodide was added to each sample to exclude labeled nonviable cells. Cells were analyzed through a LSR II flow cytometer (BD Biosciences, Mississauga, ON, Canada) set to detect yellow fluorescent protein, propidium iodide, and Alexa Fluor 647 nm in distinct channels.
For agonist-promoted endocytosis experiments, transiently transfected cells were incubated with 100 nM NDP-α-MSH, and cell surface expression was monitored after different times of agonist exposure (30 min and 1, 2, 4, 6, 8, and 22 h).
cAMP Assay.
Intracellular cAMP accumulation was measured using a competitive immunoassay based on homogeneous time-resolved fluorescence technology (cAMP dynamic-2; Cis-Bio, Bedford, MA).
Each double-tagged construct was transiently transfected into HEK293T cells. Thirty-six hours after transfection, cells were treated in the presence or absence of 10 μM antagonist for 12 h. Cells were then collected and washed (1× D-PBS, pH 7.4, and 0.1% glucose). Then 4 × 104 cells/well were dispensed in 96-well plates in cAMP buffer [1× D-PBS, 1% BSA, 0.1% glucose, 0.75 mM 3-isobutyl-1-methylxanthine (Sigma-Aldrich, Oakville, ON, Canada)] and incubated for 15 min at 37°C in the presence of 100 nM NDP-α-MSH (Sigma-Aldricht, Oakville, ON, Canada). Next, 104 cells were transferred in 384-well plates, lysed, and incubated with cAMP labeled with the dye d2 and anti-cAMP M-Antibody labeled with Cryptate according to the manufacturer's protocol. Reading of the homogeneous time-resolved fluorescence signal was performed on an Artemis time-resolved fluorescence resonance energy transfer plate reader (Cosmo Bio USA, Carlsbad, CA).
Affinity for Melanocortin Receptors.
The affinities (IC50 values) of DCPMP for recombinant human MC1R, MC3R, MC4R, and MC5R were determined at MDS Pharma Services using traditional radioligand displacement assays at the tracer concentrations indicated in Table 6.
Plasma and Brain Quantitation of DCPMP in C57BL/6 Mice.
All in vivo procedures were conducted at Eurofins Product Safety Laboratories (Des Moines, IA) under protocols approved by their Institutional Animal Care and Use Committee and followed all relevant guidance and regulation, including that set out by the Institute of Laboratory Animal Resources (1996). One of two doses (3 or 30 mg/kg) of DCPMP, formulated in 90% cottonseed oil/10% EtOH, was administered to 8-week-old C57BL/6 mice by a single intraperitoneal injection. Conventional liquid chromatography tandem mass spectrometry was used to achieve separation and detection of analytes in plasma and brain tissue (see Supplemental Experimental Procedures for description).
The elimination rate constant (kel) was calculated from the formula Ln[(Cp/2) − (1/Cp)] = −kel × t1/2 → kel = (0.693/t1/2), where t1/2 = [t2 − t1]. The half-life (t1/2) is the time taken for the plasma or brain concentration of DCPMP to fall to half its highest value. Cp is the highest concentration of DCPMP found in plasma or brain at t1 and Cp/2 is the concentration one half-life later at t2.
Statistical Analyses.
All curve fitting was conducted by nonlinear regression analyses using Prism (ver. 4.0c; GraphPad Software, San Diego, CA). Data are presented as mean ± S.E.M., and statistical significance of the differences were assessed by one-way analysis of variance. Pair-wise comparisons were made by post hoc Bonferroni's multiple comparison test. Differences with P < 0.05 were considered statistically significant.
Results
Selection and Characterization of hMC4R Mutants for PC Rescue.
Ten naturally occurring MC4R point mutations (S58C, E61K, N62S I69T, I125K, T162I, R165Q, R165W, C271Y, and P299H) that result in severe early-onset obesity in humans were used to investigate the ability of five MC4R antagonists to act as PCs. The selected mutations were chosen based on previous reports suggesting their intracellular retention (Farooqi et al., 2000, 2003; Dubern et al., 2001; Nijenhuis et al., 2003; Lubrano-Berthelier et al., 2006; Tan et al., 2009), prevalence in patient populations, and distribution throughout the receptor structural domains (Fig. 1 and Supplemental Table 1). Wild type (WT) and each of the 10 mutant receptors was tagged with a 3×HA epitope at the N terminus and a vYFP at the C terminus. Relative cell surface expression was then assessed by differentially monitoring cell surface HA immunoreactivity and total cellular vYFP fluorescence with the use of dual-flow cytometry. For the WT receptor, a generally linear relationship was seen between the total and the cell surface expression (Supplemental Fig. 1A, left). In contrast, cell surface expression of mutant receptors increased only minimally as a result of their intracellular retention until total receptor expression reached very high levels (Supplemental Fig. 1A, right). Then, to limit artifacts caused by high receptor expression (bypassing the quality control system), our analysis was limited to cells expressing low levels of receptor .The ratio between HA immunoreactivity and the vYFP signal for each individual cell was used as an index for cell surface trafficking efficiency, which has been arbitrarily fixed at 100% for WT-hMC4R and used as a reference value.
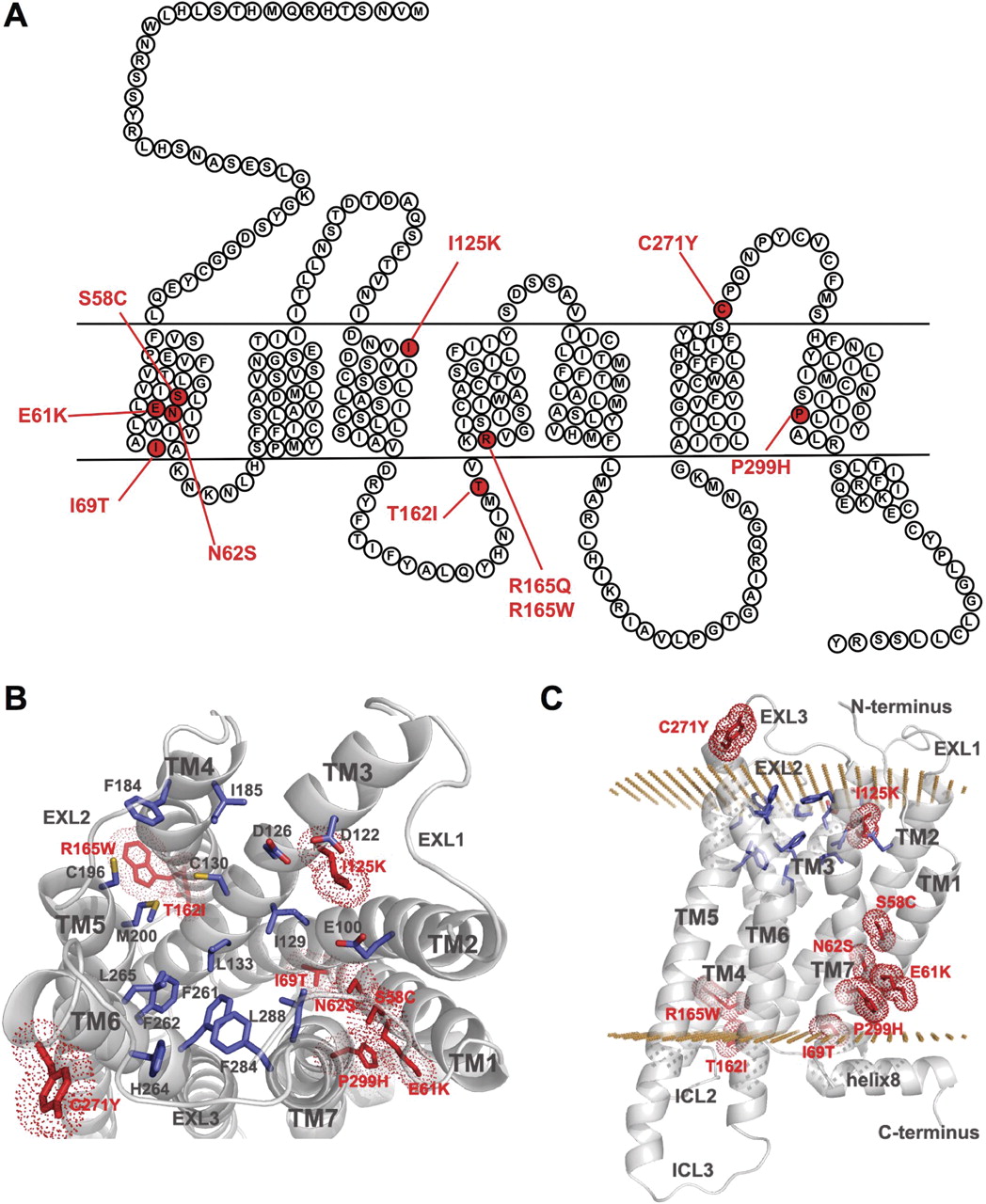
Naturally occurring mutations in hMC4R selected in this study. Schematic representation of hMC4R with the location of the 10 naturally occurring mutations selected in the study (A) and cartoon representation of the homology model of MC4R based on the β2-adrenoreceptor structural template: top view (B) and side view (C). Mutated residues are colored red, residues in the ligand-binding pocket are colored blue, and receptor helices are colored gray. Position of hydrophobic membrane boundaries is shown in accordance with the OPM database (http://opm.phar.umich.edu). Figure was prepared using PyMOL (http://www.pymol.org).
Under basal conditions, nine mutant receptors (S58C, E61K, N62S, I69T, T162I, R165Q, R165W, C271Y, and P299H) showed high levels of intracellular retention, displaying trafficking efficiencies ranging from 20 to 40% of those measured for WT receptor (Fig. 2, top). Contrary to previous reports (Yeo et al., 2003; Xiang et al., 2006), I125K-hMC4R was expressed at the cell surface to the same extent as the WT receptor (Fig. 2, top). The reason for the difference between the two studies is unclear. It should be noted, however, that despite the apparent lack of cell surface expression reported by Xiang et al. (2006) using fluorescence-activated cell sorting analysis, some binding of the cell impermeable 125I-NDP-α-MSH was observed by those authors, as well as by Yeo et al. (2003).

Characterization of cell surface expression, total expression levels, and signaling capacity of 10 mutant forms of hMC4R. Top, cell surface and total expression of 3HA-MC4R-vYFP mutants measured by fluorescence-activated cell sorting. vYFP emission represents MC4R total expression, and HA-Alexa Fluor 647 emission represents MC4R plasma membrane expression. The relative cell surface expression (ratio Alexa/vYFP emission) is calculated for each individual cell in the selected gate (see Supplemental Fig. 1) and is expressed as a percentage of the value obtained in the same experiment for the WT-hMC4R in the untreated condition. The total expression (inset) is calculated as the percentage of the mean of vYFP signal emission of each individual cell in the gate of interest (see dot-plot graphs in Supplemental Fig. 1) and is expressed as a percentage of the value obtained in the same experiment for the WT-hMC4R in untreated condition. Bottom, NDP-α-MSH-induced cAMP accumulation expressed as the percentage of agonist stimulated cAMP production of WT-hMC4R. WT-hMC4R absolute values are basal, 40 ± 8 fmol/104 cells; NDP-α-MSH-stimulated cAMP production, 300 ± 20 fmol/104 cells. Each bar represents the mean ± S.E.M. of at least three independent experiments performed in triplicate.
It is noteworthy that the reduced cell surface trafficking of the nine other mutants was not accompanied by significant changes in their total expression levels, as illustrated by their equivalent average vYFP signals (Fig. 2, top, inset). To investigate the signaling capacity of the mutant receptors, cAMP accumulation was determined after agonist stimulation with 100 nM NDP-α-MSH. In contrast to the WT receptor, NDP-α-MSH did not significantly stimulate cAMP production for any of the hMC4R mutants (Fig. 2, bottom).
Selection of MC4R Compounds and Docking in the hMC4R Model.
Five MC4R antagonists with distinct structural features (Table 1) were selected as potential PCs based on published high affinity and selectivity for MC4R and their predicted lipophilic properties, allowing penetration into intracellular compartments. The putative binding modes of these compounds on WT-hMC4R were assessed by virtual docking and are presented in Supplemental Fig. 2, and the major contact points are listed in Table 2.
Antagonists selected in the study
Values were obtained from radioligand displacement assays by competition with 0.02 nM 125I-NDP-α-MSH on HEK293 cells expressing hMC4R (Kd,, 0.5 nM; Bmax, 3900 fmol/mg protein).
Description of binding residues for THIQ and antagonists of the study
Residues of MC4R located at 4.5 or 4.0 Å from PCs docked in the models of MC4R receptor in the inactive conformation. Residues that are important for THIQ binding (Protein Data Bank code 2IQU) are in bold. Residues presented in Supplemental Fig. 2 are underlined.
Low-energy conformations of all five MC4R-selective antagonists were docked into the binding pocket of the hMC4R inactive state model, based on structure-activity relationship (SAR) studies (Arasasingham et al., 2003; Pontillo et al., 2005a,b; Vos et al., 2006), knowledge of functionally important receptor residues (Pogozheva et al., 2005; Yang et al., 2009), and in accordance with geometric and polarity matching of ligand and receptor (Table 2 and Supplemental Fig. 2).
Three of the antagonists (PPPone, DCPMP, and NBP) could be docked similarly to the well characterized hMC4R-selective small-molecule agonist THIQ (Sebhat et al., 2002) (Table 2 and Supplemental Fig. 2A). DCPMP is structurally related to THIQ, harboring a central halogen-substituted 2,4-Cl-d-Phe aromatic ring (“A”) and a phenylpiperazine moiety (“B”) that mimics the cyclohexylpiperazine of THIQ (Table 2 and Supplemental Fig. 2E). The basic benzylamine group of DCPMP replaces the N-terminal basic nitrogen of THIQ. The two dipiperazine-based ligands, PPPone and NBP, also have central halogen-substituted phenyl rings (“A”), and a second aromatic function (“B”) that are essential for high binding affinity (Arasasingham et al., 2003), as well as basic nitrogens in the piperazinyl rings. The central halogen-substituted aromatic ring of these three ligands (“A”) represents the key pharmacophore and can occupy the same position at the bottom of the binding cavity as the p-chlorophenyl ring of THIQ, forming multiple contacts with aromatic, aliphatic, and sulfur-containing residues from TM3 (Ile129, Cys130, Leu133), TM5 (Cys196, Met200), TM6 (Phe261, Leu265), and TM7 (Phe284, Leu288). At physiological pH, the benzylamine group in DCPMP (pKa = 8.97) and the central piperazinyl group in NBP (pKa = 8.92) are positively charged and can form ionic pairs with Asp126 in TM3 (Table 2 and Supplemental Fig. 2, E and F). Furthermore, the nitrogens in the terminal piperazinyl groups of PPPone (pKa = 8.21) and NBP (pKa = 7.91) are also positively charged and may be involved in ionic interactions with both Asp126 (TM3) and Glu100 (TM2) (Table 2 and Supplemental Fig. 2, C and F). The nitrogen from the central piperazinyl group of PPPone (pKa = 7.48) would be charged only at a more acidic pH. Additional hydrophobic interactions may form between the benzyl rings of DCPMP, PPPone, or the naphthyl ring of NBP and residues from TM4 (Phe184, Ile185), TM5 (Cys196, Met200), and TM6 (Leu265), similar to interactions of the cyclohexyl group and the triazol ring of THIQ. The benzyl ring of DCPMP extends toward the extracellular surface, where it may interact with residues from TM4 (Phe184, Ile185) as well as with residues from extracellular loop (EXL) 2 and the N terminus.
Docking of the structurally distinct ligands MPCI and MTHP was different from that proposed for PPPone, DCPMP, and NBP. SAR studies of MPCI indicate the functional importance of the basic group in the arylguanidine substituent in combination with the central fluorobenzyl ring (“A”) and the second aromatic ring (“B”) with lower lipophilicity. Therefore, we propose that the low-energy conformations of MPCI may be docked such that the N+ of the piperidinyl group forms an ion pair with Glu100 (TM2) and Asp126 (TM3), whereas the fluoro-benzyl ring occupies a position similar to the 4-chlorophenyl ring of THIQ (Table 2 and Supplemental Fig. 2D). The docking of MTHP is more challenging. A folded conformation of the ligand was chosen, because this is energetically preferred compared with an extended conformation (ΔE > 3.5 kcal/mol). Because of the small size, two molecules of MTHP can be easily accommodated within the relatively large receptor binding pocket: one molecule occupying the area between TMs 3 and 6 and another molecule filling the space between TMs 2, 3, and 7. The positively charged nitrogen of the ligand can form an ionic pair with Asp126 (TM3) if the ligand is in the first docking pose or with Glu100 (TM2) if the ligand occupies the second docking pose (Table 2 and Supplemental Fig. 2B).
Cell Surface Rescue of Mutant hMC4Rs.
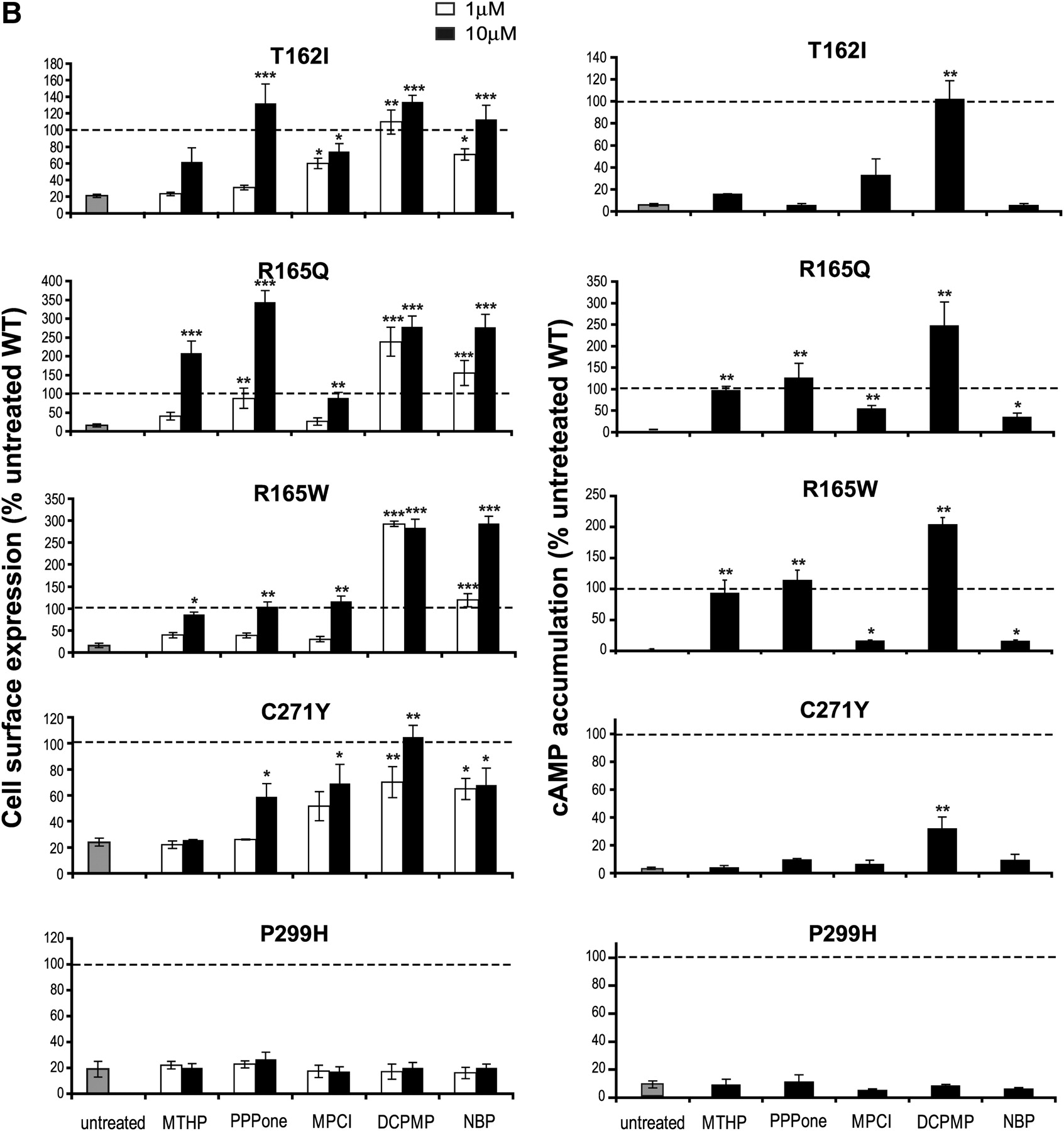
The ability of the selected MC4R antagonists to act as PCs was first assessed by monitoring receptor cell surface expression after 12-h incubation with the compounds at 1 and 10 μM. Quantitative analysis of the flow cytometry dot plots (Supplemental Fig. 1 for WT and I69T hMC4R) revealed that cell surface expression of all mutant receptors, except for P299H, was significantly increased after incubation with at least one of the compounds (Fig. 3, left). It is noteworthy that the efficiency of the individual compounds to promote cell surface expression was mutation-dependent.

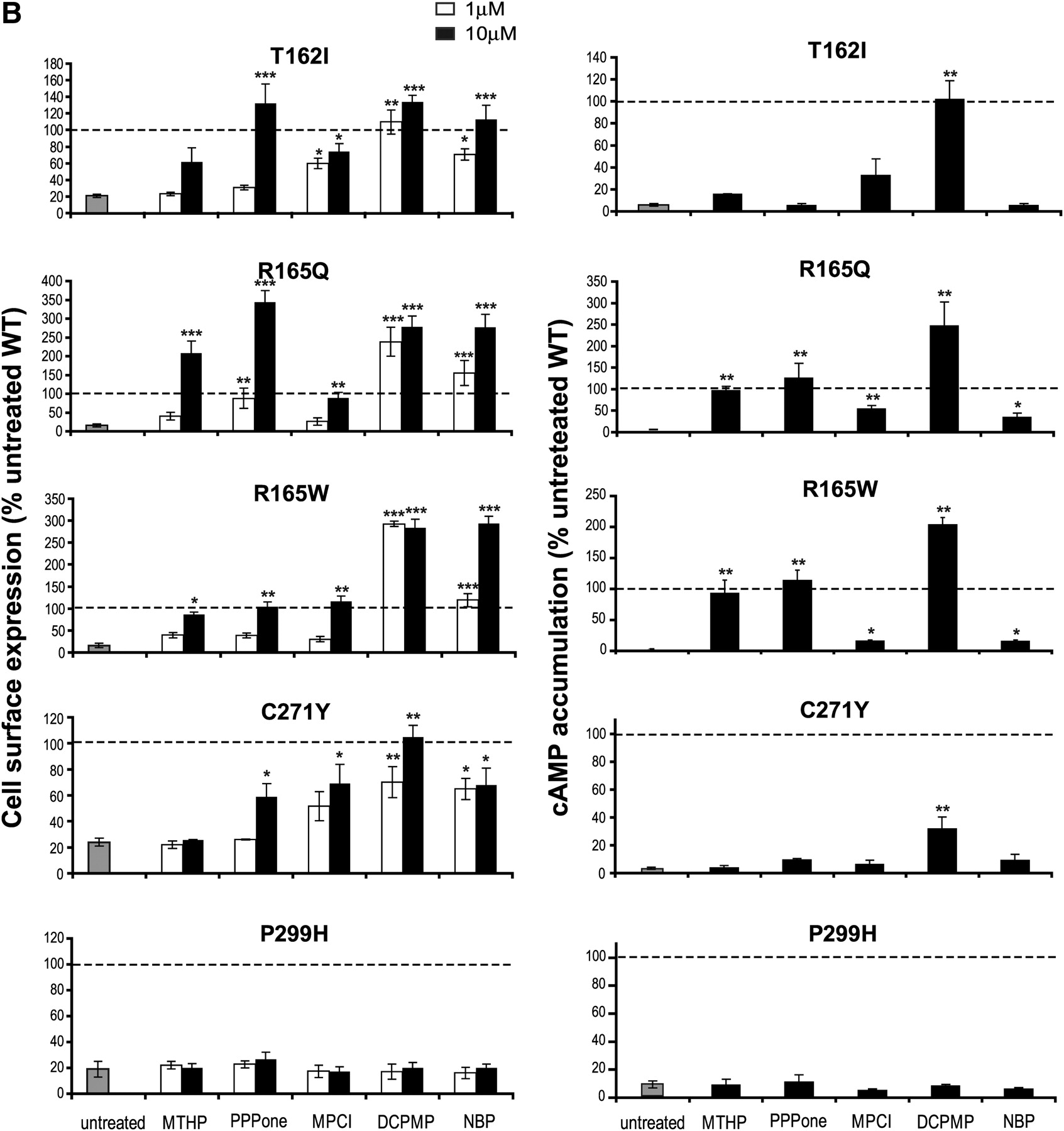
Effects of MC4R antagonists on cell surface expression and stimulated cAMP production of mutant and WT hMC4Rs. HEK293T cells transiently expressing the indicated double-tagged hMC4R were incubated for 12 h in the absence (untreated) or presence of antagonist compounds. Left, cell surface expression of each receptor was measured by flow cytometry. Cells were incubated in the absence (gray bar, untreated) or presence of 1 μM (open bar) or 10 μM (filled bar) antagonist compounds. The relative membrane expression (ratio of Alexa/vYFP emission) was calculated for each individual cell in the gate of interest (see dot-plot graphs in Supplemental Fig. 1) and expressed as a percentage of the value obtained in the same experiment for the WT-hMC4R in the absence of antagonist. Right, HEK293T cells transiently expressing the indicated double-tagged hMC4R were incubated for 12 h in the absence (gray bar, untreated) or presence (filled bar) of 10 μM antagonist compounds. cAMP accumulation was measured after 100 nM NDP-α-MSH stimulation. The results are expressed as the percentage of NDP-MSH-stimulated cAMP production by WT-hMC4R. Each bar represents the mean ± S.E.M. of at least three independent experiments. Dashed lines represent WT-hMC4R cell surface expression (left) or cAMP accumulation (right) in untreated condition. This level is considered as reference for a full recovery for mutant receptors. The symbol (*) indicates a significant difference from untreated condition: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
At the highest concentration tested, DCPMP and NBP showed the broadest activity, significantly increasing the cell surface targeting of all nine responsive mutants (Fig. 3, left and Table 3). PPPone increased cell surface expression of the same subset of mutant receptors, except for I125K. MTHP significantly increased cell surface levels of S58C, I125K, R165Q, and R165W, whereas MPCI enhanced cell surface expression of T162I, R165Q, R165W, and C271Y. In some cases, PCs increased cell surface expression at higher levels than that of untreated WT receptor (e.g., DCPMP and NBP for S58C, I125K, R165Q, and R165W; MTHP for I125K and R165Q; PPPone for I69T and R165Q).
Overview of efficacy and relative potency of each compound on cell surface rescue
Untreated indicates relative membrane expression reported as a percentage of the value obtained in the same experiment for the hMC4R (WT) in the absence of antagonist. Efficacy refers to relative surface expression level attained after overnight incubation with 10 μM antagonist. Relative potency is the ratio between the mean of the relative surface expression level obtained at 1 and 10 μM. Data shown are the mean ± S.E.M. of at least three independent experiments and are derived from Fig. 3.
The relative apparent potency of the compounds to promote cell surface targeting also varied among the different mutants. This is evident based on the efficiency ratios of cell surface rescue promoted by incubation with the different compounds at 1 and 10 μM (Table 3). DCPMP and NBP showed high relative potencies for promoting cell surface targeting (ratios >0.6) of eight of nine and six of nine mutant receptors, respectively. In contrast, MTHP and PPPone showed lower relative potencies (ratios <0.6) for four of four and eight of eight of the rescued mutants, respectively. Finally, MPCI showed an intermediary relative potency profile with half of the four rescuable mutants below and half above the 0.6 ratio. All compounds had potency ratios below 0.6 to rescue N62S, as well as low potencies to rescue R165W and R165Q, except DCPMP. In contrast, T162I and C271Y were rescued with potency ratios above 0.6 by three of the five compounds: MPCI, DCPMP, and NBP. It should be noted that although the relative potency value allows a comparison of the potencies when the ratio is below 1, it does not allow a conclusion that compounds that have ratios of 1 have identical potencies, because such a ratio simply indicates that the potency of the compound is below 1 μM.
Although the compounds varied significantly in their ability to rescue the cell surface expression of different mutants, some generalities can be drawn: two mutants (R165Q and R165W) are rescued by all compounds, two compounds (DCPMP and NBP) rescue all mutants, and the subset of mutations that could be partially or fully rescued is different for each compound (Table 3). It is noteworthy that MTHP, DCPMP, and NBP also significantly increased the relative cell surface expression of the WT receptor with high potency (relative potency ratios of 0.74, 0.91, and 0.96, respectively), indicating that trafficking efficiency of WT-hMC4R could also be influenced by PCs.
Functional Rescue of hMC4R Mutants with PCs.
We next investigated whether PC-mediated increases in cell surface expression could also restore signaling activity of the mutant MC4Rs (Fig. 3, right). In the case of I125K, none of the compounds could restore NDP-α-MSH-stimulated cAMP response, despite the fact that its cell surface expression could be further increased by three compounds (MTHP, DCPMP, and NBP), indicating that this mutant form of the receptor is unable to respond to agonist. This exception aside, compounds that increased cell surface expression also restored NDP-α-MSH-stimulated cAMP production (Fig. 3, right) for all mutants, confirming that rescued mutant receptors are in a conformation that can bind agonist and transduce signal. However, differences in the extent of cell surface and functional rescue were observed. For instance, although NBP very efficiently rescued the cell surface expression of six mutants, it only marginally restored significant signaling for two of them. This most likely results from persistent NBP binding because of its high affinity (2 nM), which prevents subsequent stimulation with NDP-α-MSH. This hypothesis is supported by the observation that pretreatment of WT-hMC4R with NBP led to a ∼50% inhibition of NDP-α-MSH-stimulated cAMP production (Fig. 3, right).
Even for compounds that have lower affinity for the receptor, discrepancies between rescued cell surface expression and signaling activity were observed. This is illustrated by the signaling/cell surface ratio, which is taken as an indication of the signaling efficacy of the rescued receptor and is set at one for the WT-hMC4R (Table 4). For four of the mutants (I69T, T162I, R165Q, and R165W), at least one of the compounds restored signaling efficacy ratios to values above 0.8. For other mutations (S58C, E61K, N62S, and C271Y), none of the tested compounds promoted signaling ratios above 0.8, indicating that the conformation stabilized for these mutants may be less efficient for signaling. It is noteworthy that when considering each mutant receptor the compounds that favored the highest signaling efficacy ratios were not always the same. For example, DCPMP promoted the highest signaling ratios for R165Q, T162I, and E61K, whereas MTHP and PPPone did so for R165W and I69T.
Comparison of cell surface expression and signaling efficacy for each compound
Cells expressing WT or mutant hMC4Rs were preincubated in the absence or presence of 10 μM concentrations of the indicated compound for 12 h. After washes, cells expressing WT or mutant hMC4Rs were stimulated for 15 min at 37°C with 100 nM NDP-α-MSH.
In several cases, incubation with specific compounds restored cAMP responses that were equivalent or superior to those observed for untreated WT receptor (Fig. 3, right). Such complete rescue was seen for different compound/mutation combinations (MTHP, R165Q and R165W; PPPone, I69T, R165Q, and R165W; DCPMP, S58C, E61K, N62S, I69T, T162I, R165Q, and R165W). Overall, DCPMP emerged as the compound that restored function to the largest subset of mutants. As was the case for cell surface expression, incubation of WT receptor with MTHP, PPPone, or DCPMP also increased NDP-α-MSH-stimulated cAMP production.
Pharmacological Characterization of Rescued Receptors.
To further explore the pharmacological properties of the rescued receptor, we generated concentration-response curves for NDP-α-MSH- and α-MSH-stimulated cAMP production in cells expressing WT or three MC4R mutants selected for their different rescue profiles (R165W, N62S, and C271Y; Table 5 and Supplemental Fig. 3). Treatment of cells expressing the WT receptor with DCPMP did not affect the EC50 of either hormone to stimulate cAMP production. For DCPMP-rescued R165W, the potency of NDP-αMSH and α-MSH was almost identical or slightly reduced compared with WT (1.2- and 4-fold, respectively) (Table 5). The potency of both agonists for the rescued N62S was reduced by approximately 5-fold compared with WT receptor. These data indicate that the DCPMP-rescued R165W and N62S have signaling potencies that are not drastically different from those of the WT receptor. In contrast, the EC50 values for both NDP-α-MSH- and α-MSH-stimulated cAMP production for the DCPMP-rescued C271Y were much higher (44- and 32-fold, respectively) than for WT receptor, indicating a significant loss of potency for this receptor mutant (Table 5).
Efficacy and potency of melanocortin agonists to stimulate cAMP production of WT and mutant hMC4Rs rescued by DCPMP pretreatment
Data shown are the mean ± S.E.M. of the number of experiments indicated in parentheses. EC50 is the concentration of ligand that results in 50% stimulation of the maximal response (Rmax).
Pharmacological Characterization of DCPMP.
Because DCPMP efficiently rescued the largest subset of MC4R mutants, it emerges as a potential lead for the development of a therapeutic PC to treat MC4R-linked obesity. Thus, its potency, selectivity, and in vivo pharmacokinetic profile were assessed. The potency of DCPMP to functionally rescue the NDP-α-MSH-stimulated cAMP production by N62S, E61K, and R165W is illustrated in Fig. 4. In agreement with the weaker relative potencies of DCPMP to restore cell surface expression of N62S (0.52) and E61K (0.61) versus R165W (1.04) (see Table 3), the dose-response curve of DCPMP to restore signaling activity was right-shifted for N62S and E61K compared with R165W. The lack of saturation of the responses for N62S and E61K prevented the determination of an accurate EC50, but an EC50 of 1.5 μM was calculated for R165W. This value is 10-fold higher than that obtained for the WT receptor to enhance the signaling response above untreated condition level (data not shown). Although no direct information regarding the affinity of DCPMP for the mutant form of the receptor is available [i.e., its relative potency close to 1 for both WT and R165W (Table 3) indicates that its potency is better than 1 μM but not necessarily equal for the two receptor forms], the difference in its potency to promote R165W versus WT signaling could indicate that the mutation affects the affinity of the receptor for DCPMP. Alternatively, the persistence of the rescued mutant receptor functionality could be reduced compared with the WT receptor as a consequence of an accelerated loss of cell surface receptor upon activation. Thus, a higher concentration of the PC may be needed to reach an equivalent functional steady state than with the WT receptor. To compare the kinetics of cell surface residency of the rescued R165W with that of the WT receptor, we assessed the kinetics of agonist-promoted endocytosis of the R165W and WT receptors after a 12-h pretreatment with DCPMP and compared it with that of the untreated WT receptor. As shown in Fig. 5, stimulation of the receptors with the agonist NDP-α-MSH led to a loss of cell surface receptors with comparable kinetics for the rescued R165W and the WT receptor (pretreated or not with DCPMP), indicating that the dynamics of cell-rescued R165W once at the cell surface are identical to those of the WT receptor. It is therefore unlikely that the difference in the apparent potency of DCPMP may result from distinct kinetics of cell surface residency of the rescued receptor.

Concentration-response curve of DCPMP treatment on mutant forms of hMC4R. Signaling capacity of E61K-, N62S- and R165W-MC4R upon stimulation with 100 nM NDP-α-MSH, after 12-h incubation with increasing concentrations of DCPMP. The data are expressed as the percentage of maximal level of cAMP accumulation of mutant forms of hMC4R upon agonist stimulation in the presence of DCPMP preincubation. Each point represents the mean ± S.D. of two to three independent experiments performed in triplicate.
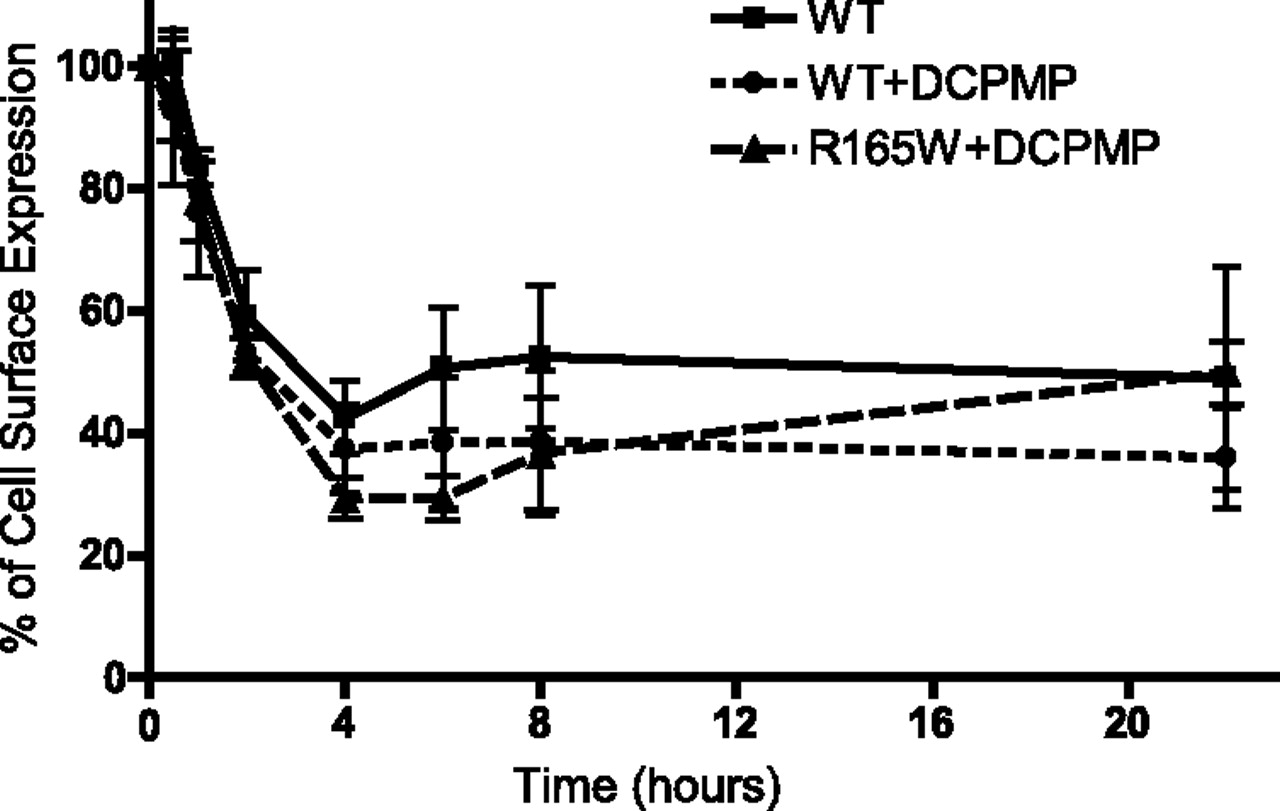
Kinetic of agonist-promoted endocytosis of rescued R165W- and WT-MC4R. HEK293T cells transiently expressing R165W and WT double-tagged hMC4R were incubated for 12 h in the absence (only for WT-MC4R) or presence of 10 μM DCPMP. Agonist-promoted endocytosis was induced by 100 nM NDP-α-MSH maintained over time. The cell surface expression level was measured by flow cytometry after incubation with agonist for 30 min and 1, 2, 4, 6, 8, and 22 h. The cell surface expression level is expressed as the percentage of the value measured at T0 (no agonist stimulation) in each condition. Each point represents the mean ± S.D. of three independent experiments.
The selectivity of DCPMP for MC4R was assessed by its ability to inhibit 125I-NDP-α-MSH binding to human MC1R, MC3R, MC4R, and MC5R. DCPMP displayed high affinity for MC4R with an IC50 value of 25 nM, showing a selectivity of 100-fold or more over the three other receptor subtypes (Table 6).
Melanocortin receptor selectivity of DCPMP
The affinities (IC50 values) of DCPMP for recombinant human MC1R, MC3R, MC4R, and MC5R in HEK293 cells were determined using traditional radioligand displacement assays in the presence of 0.04 nM 125I-NDP-α-MSH for MC1R (Kd, 0.037 nM); 0.035 nM 125I-NDP-α-MSH for MC3R (Kd, 0.24 nM); 0.02 nM 125I-NDP-α-MSH for MC4R (Kd, 0.5 nM); 0.035 nM 125I-NDP-α-MSH for MC5R (Kd, 0.53 nM).
To determine whether DCPMP could reach its intended target in the central nervous system, one of two doses of DCPMP (3 and 30 mg/kg) were administered to 8-week-old C57BL/6 mice by a single intraperitoneal injection. The presence of compound was then monitored over a 24-h period in blood and brain (Fig. 6). DCPMP showed similar kinetics in these two tissues, reaching maximal concentrations at 30 and 60 min after injection for the 3 and 30 mg/kg dose groups, respectively. Although the maximal concentrations reached in the brain were 16- and 8-fold lower than those in the plasma, the level reached after 30 mg/kg administration (0.5 μM) is very close to the EC50 for PC activity determined in cells (see Fig. 4). The calculated elimination rate constants were similar for brain and plasma (3 mg/kg: 0.69 ng · ml−1 · h−1 for plasma and brain; 30 mg/kg: 0.35 ng · ml−1 · h−1 for plasma and 0.23 ng · ml−1 · h−1 for brain). It is noteworthy that more than 80% of the drug was cleared from brain 4 and 8 h after injection for the 3 and 30 mg/kg dose groups, respectively.

Pharmacokinetic profile of the MC4R antagonist DCPMP. One of two doses of DCPMP [3 mg/kg (square) and 30 mg/kg (triangle)] were administered to 8-week-old C57BL/6 mice by a single intraperitoneal injection. DCPMP concentrations in blood and brain were measured at the indicated time. The dashed and solid lines correspond to the concentration measured in brain and blood, respectively. To derive approximate molar concentrations of DCPMP in the brain, we assumed that 1 g of tissue corresponds to a volume of 1 ml.
Discussion
Our study revealed important mutation-specific differences in the PC action of MC4R antagonists that have distinct chemical structures. Nevertheless, at least one PC rescued almost every MC4R mutant tested. Indeed, only 2 of the 10 MC4R mutants were resistant to PC treatment: P299H, the cell surface expression of which could not be restored by any PC, and I125K, which had normal cell surface expression but could not be functionally rescued by any compound despite a facilitation of its cell surface targeting by some PCs. The P299H mutation affects the proline from the conserved N/DPxxY motif and is predicted to eliminate the proline-induced kink in TM7, affecting the packing of TM1, TM2, and TM7 (Tan et al., 2009). This conformational change might result in an unstable receptor that is most likely unable to bind PCs. The lack of functional response observed for I125K is consistent with the large decrease in binding affinity of NDP-α-MSH that was reported previously (Haskell-Luevano et al., 2001; Yeo et al., 2003; Chen et al., 2007). However, this mutation did not prevent the binding of all ligands; MTHP, DCPMP, and NBP potentiated its cell surface expression. Despite this increased cell surface expression, no signaling activity was restored by these three PCs, indicating that the conformations stabilized by the PCs are unable to bind NDP-α-MSH with high affinity and/or are unable to transduce the binding into signaling.
Among the five PCs tested, four were able to rescue signaling activity of at least two of the eight rescuable mutants. The only compound that could not restore significant NDP-α-MSH responsiveness was NBP, despite a very potent and efficacious rescue of the cell surface trafficking, most likely as a consequence of prolonged antagonist action resulting from its very high affinity. The presence of the bulky naphthyl ring as well as two positively charged N+ from both piperazinyl groups that form multiple interactions with receptor residues (Table 2 and Supplemental Fig. 2) may explain the higher binding affinity and slow dissociation rate of this ligand.
When considering the four compounds that rescued function, distinct mutation-dependent PC efficacy was observed. Whereas DCPMP significantly restored function to all rescuable mutants, the signaling activity of only some were significantly rescued by PPPone (N62S, I69T, R165Q, and R165W), MTHP (I69T, R165Q, and R165W), and MPCI (R165Q and R165W). The broader PC action of DCPMP cannot be predicted simply on the basis of the affinity of the ligand for the WT-hMC4R. Indeed, PPPone, which rescues the second largest number of mutant receptor forms, has the lowest affinity. This suggests that structural effects of the mutation as well as the binding modes of the compounds must contribute to the mutant-specific profiles of the PCs. For example, DCPMP restored the highest efficacy ratios for T162I (0.76) and R165Q (0.9), whereas PPPone promoted efficacy ratios of only 0.05 and 0.37 for the same mutants. In contrast, PPPone restored a higher signaling efficacy than DCPMP for I69T (0.96 versus 0.66). This mutant-dependent action of the PCs may be explained by the fact that Thr162 and Arg165 are at the bottom of TM4, a TM that is predicted by our docking data (Table 2 and Supplemental Fig. 2, C and E) to have more contacts with DCPMP than with PPPone. Thus, the interactions of DCPMP with Phe184 and Ile185 at the top of TM4 may compensate for the destabilization of the H-bond network between TM4 and the bottom of TM3 and intracellular loop 2 by maintaining the packing of TM4 in the receptor bundle. On the other hand, unlike DCPMP, PPPone interacts with residues of TM2 (Glu100, Ile104), interactions that may stabilize the helix packing between TM2 and TM1, which could have been disrupted by the substitution of the hydrophobic Ile69 with the smaller polar threonine. It is noteworthy that the most broadly active compounds (DCPMP, NBP, and PPPone) share a common general binding mode that involves a greater number of receptor residues (Table 2 and Supplemental Fig. 2), indicating that the multiple interactions formed by these three piperazine-containing compounds can stabilize receptors harboring conformational defects originating from mutations in different receptor regions. Despite this general rule, the cell surface expression of two mutants, E61K and C271Y, could be restored to WT levels by only one PC (DCPMP). This may be explained by the more dramatic conformational changes predicted to result from these mutations, making them more resistant than others to stabilization by most PCs. Indeed, substitution of the TM1 Glu61 by a lysine residue has been proposed to promote the formation of an aberrant hydrogen bond with the free carbonyl group of Ile296 in TM7 that may change packing interactions of TM1 and TM7 (Tan et al., 2009). The C271Y substitution prevents the formation of a disulfide bond (Cys271–Cys277) between the end of TM6 and EXL3 (Tarnow et al., 2003).
When considering signaling efficacy in light of the cell surface rescue, two general scenarios were observed: the signaling efficacy was either proportional or significantly less than predicted from the cell surface targeting. The first scenario is exemplified by I69T, T162I, R165Q, and R165W, for which some PCs restored the intrinsic signaling efficacy to levels similar to or above those of untreated WT, indicating that PC treatments promoted conformations that are fully competent for signaling. This is somewhat surprising for R165Q and R165W, because this arginine has been proposed to interact with Asp146 of the DRY motif and thus to be important for ligand-promoted MC4R activation (Xiang et al., 2006). Our data indicate that, although the Arg165 residue may be important in promoting a specific conformation, it is not essential for receptor activation, as confirmed by the recovery of the same agonist's potency as the WT receptor in functional assay after rescue with DCPMP (Table 5). The second scenario is exemplified by S58C, E61K, N62S, and C271Y, for which none of the PCs could restore signaling efficacies above 0.75 (see Table 4). Mutations S58C, E61K, and N62S are predicted to alter the H-bonding interactions between polar residues from TM1 (Ser58 and Asn62), TM2 (Asp90, Ser94, and Asn97), and TM7 (Asn294, Ser295, and Asp298), which play a central role in receptor activation (Govaerts et al., 2005; Tan et al., 2009). For C271Y, the loss of the disulfide bond in EXL3 most likely results in a major conformational change that could alter NDP-α-MSH binding or the translation of ligand binding into receptor activation. Consistent with this notion, the potencies of NDP-α-MSH and α-MSH to stimulate cAMP production for the DCPMP-rescued C271Y-MC4R were decreased by 44- and 32-fold, respectively, compared with WT receptor (Table 5).
The different compounds also had different relative potencies and efficacies to promote cell surface expression and enhance cell signaling of WT-hMC4R (Table 4). This observation suggests that a fraction of native receptors fails to achieve proper folding and that the presence of a PC favors such folding. Similar effects of PCs have been reported previously for the δ-opioid (Petäjä-Repo et al., 2002) and GnRH (Janovick et al., 2002; Conn et al., 2007) receptors. Therefore, in addition to representing a therapeutic avenue for severely obese patients carrying MC4R mutations, PC treatment could possibly be useful for the treatment of obesity in patients with normal MC4R.
It should be noted that the ability of the PCs to restore function was assessed only for the canonical Gs-adenylyl cyclase signaling pathway. Given that it has been proposed that different ligand-promoted GPCR conformations may be responsible for the activation of distinct signaling pathways (Galandrin et al., 2007; Rajagopal et al., 2010), it will be interesting to monitor the action of various PCs on the ability of mutant MC4Rs to activate other signaling effectors, such as the recruitment of β-arrestin and the activation of MAPK.
To be clinically useful, antagonist PCs need to 1) selectively bind the targeted receptor; 2) have a sufficiently high affinity for the receptor but a rapid dissociation rate that allows washout and competition by the endogenous ligand; 3) be sufficiently lipophilic to penetrate cell membranes; and 4) be as potent and efficacious as possible on the largest subset of receptor mutant forms. DCPMP possesses many of these characteristics (e.g., high selectivity toward MC4R, broad efficacy toward many mutants, ability to reach the brain in sufficient concentration, and relatively rapid clearance), making it an interesting lead compound for the development of a therapeutically useful PC. Nevertheless, our data clearly indicate that different compounds could better rescue some mutations, suggesting that pharmacogenetics may play an important role in the establishment of PCs as therapeutics. Because the current strategies to control early-onset obesity are either modestly effective or very invasive (bariatric surgery), the development of clinically active PC represents an attractive avenue.
Acknowledgments
We thank Dr. Monique Lagacé for useful discussion and assistance in the preparation of the manuscript.
Footnotes
This work was supported by Amicus Therapeutics (to M.B.); the Canadian Institute for Health Research [Grant 10501] (to M.B.); and a Canada Research Chair in Cell Signaling and Molecular Pharmacology (to M.B.).
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.110.172098.
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- GPCR
- G protein-coupled receptor
- ER
- endoplasmic reticulum
- PC
- pharmacological chaperone
- MC4R
- melanocortin-4 receptor
- hMC4R
- human melanocortin-4 receptor
- HA
- hemagglutinin
- MTHP
- 2-(2-(2-methoxy-5-nitrobenzylthio)phenyl)-1,4,5,6-tetrahydropyrimidine
- PPPone
- 3-(4-(2-(4-fluorophenyl)-2-(4-methylpiperazin-1-yl)ethyl)piperazin-1-yl)-2-methyl-1-phenylpropan-1-one
- MPCI
- 2-(5-bromo-2-methoxyphenethyl)-N-(N-((1-ethylpiperidin-4-yl)methyl)carbamimi doyl)-3-fluorobenzamide
- DCPMP
- N-((2R)-3(2,4-dichlorophenyl)-1-(4-(2-((1-methoxypropan-2-ylamino)methyl)phenyl)piperazin-1-yl)-1-oxopropan-2-yl)propionamide
- NBP
- 1-(1-(4-fluorophenyl)-2-(4-(4-(naphthalene-1-yl)butyl)piperazin-1-yl)ethyl)-4-methylpiperazine
- PBS
- phosphate-buffered saline
- BSA
- bovine serum albumin
- NDP
- [Nle4,d-Phe7]
- α-MSH
- α-melanocyte stimulating hormone
- HEK
- human embryonic kidney
- D-PBS
- Dulbecco's PBS
- WT
- wild type
- vYFP
- venus yellow fluorescent protein
- SAR
- structure-activity relationship
- THIQ
- tetrahydroisoquinoline
- TM
- transmembrane domain
- EXL
- extracellular loop.

- Received June 30, 2010.
- Accepted September 7, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}