Article Text

Abstract
Objective: To determine the efficacy, impact on quality of life (QOL) and safety of prucalopride, a selective, high-affinity 5-HT4 receptor agonist, in patients with chronic constipation.
Methods: In this multicentre, randomised, placebo controlled, parallel-group, phase III study, patients with chronic constipation (two or fewer spontaneous complete bowel movements (SCBM)/week) received 2 mg or 4 mg prucalopride or placebo, once daily, for 12 weeks. The primary efficacy endpoint was the proportion of patients reaching three or more SCBM/week. The key secondary efficacy endpoint was the proportion of patients having an increase of one or more SCBM/week. The primary QOL endpoint was the patient assessment of constipation QOL satisfaction subscale score. Safety parameters included adverse events, laboratory values and cardiovascular events.
Results: Efficacy was evaluated over 713 patients. Averaged over 12 weeks, higher proportions of patients on prucalopride 2 mg (19.5%; p<0.01), 4 mg (23.6%; p<0.001) had three or more SCBM/week (or normalisation of bowel function) compared with placebo (9.6%). Similar results were seen in the subgroup (83%) of patients dissatisfied with previous laxative treatment. Both doses of prucalopride also significantly improved secondary efficacy and QOL endpoints, including the proportion of patients with an increase of one or more SCBM/week, evacuation completeness, perceived disease severity and treatment effectiveness and QOL. Prucalopride 4 mg significantly reduced the need for straining versus placebo (p<0.05). The most frequent treatment-related adverse events were headache and diarrhoea. Both doses of prucalopride were safe and well tolerated.
Conclusion: Prucalopride significantly and consistently improved bowel function, associated symptoms and satisfaction in chronically constipated patients.
Trial registration number: NCT00488137.
Statistics from Altmetric.com
Constipation is a highly prevalent, often chronic, gastrointestinal disorder that mainly affects women and older adults.1 2 Prevalence estimates vary, but typically range from 10% to 15% in developed countries.3 4 In a recent survey of nearly 14 000 people, 12% reported constipation and 51% had experienced symptoms for three or more years.5
A variety of laxatives are available to individuals with constipation. Despite the widespread and long-term use of laxatives5 and general acceptance of their efficacy, clinical evidence of long-term efficacy is limited.6–8 Many constipated patients report high levels of dissatisfaction with laxative treatment and often do not obtain adequate long-term relief from laxatives.6–9 In one survey, 71–75% of patients were not satisfied with the predictability of laxative therapy, 50–67% with the inadequate relief of bloating and other symptoms and 44–50% with the lack of efficacy in relieving constipation.9 Furthermore, 44–68% of patients were not satisfied with the effect of laxatives on improving quality of life (QOL).9 10 Osmotic laxatives can have a bad taste,11 and bulk-forming laxatives require large water intakes that may be difficult for elderly people. In addition, laxatives do not address the underlying pathophysiology of constipation.
Constipation is associated with impaired lower gastrointestinal motility, often with a reduction in the giant migrating contractions that normally drive mass transits through the colon.12 More specifically, subtle alterations of the enteric nervous system (ENS) are thought to underlie motility impairment.13 14 A logical approach to treat chronic constipation therefore involves physiological stimulation of intestinal motility by targeting neurons of the ENS.12 15
Prucalopride is a selective, high-affinity agonist at serotonin 5-HT4 receptors, which are expressed by ENS neurons and through which prucalopride exerts potent enterokinetic effects.16–18 Prucalopride belongs to a novel chemical class; it is a dihydrobenzofurancarboxamide derivative and is therefore structurally different from other serotonergic prokinetic agents such as cisapride and tegaserod. These agents are less selective than prucalopride for 5-HT4 receptors and, in the same concentration range, interact with other receptors such as 5-HT3, 5-HT1B/D and the hERG potassium channel.15 19–21 Prucalopride is eliminated without extensive metabolism. Therefore, prucalopride has a low potential for drug–drug interactions and the coadministration of drugs that inhibit CYP450 will have no clinically relevant effect on prucalopride plasma concentrations (Van de Velde, Ausma, Vandeplassche, UEGW, 2008, unpublished observations).
Previous clinical studies (phase I to III) have shown that prucalopride significantly improves colon motility and transit, the frequency of bowel movements and patient satisfaction with bowel function.22–26
The objectives of this phase III placebo controlled trial were to determine the efficacy, safety and impact on QOL, of oral prucalopride (2 mg and 4 mg), given once daily, for 12 weeks in patients with chronic constipation.
METHODS
Study design
This international, multicentre, randomised, double-blind, placebo controlled, parallel-group trial was conducted in seven countries from 13 March 1998 to 19 July 1999. Two factors have contributed to the delay between data collection and submission of this study for publication: the transfer of prucalopride (and other assets) from Johnson & Johnson to Movetis between 2003 and 2006 and the compilation of an extensive safety and toxicology package between 1999 and 2003.
A 12-week treatment period was preceded by a 2-week run-in phase (baseline). Eligible patients with less than two spontaneous complete bowel movements (SCBM) per week were randomly assigned to 2 mg or 4 mg of prucalopride or placebo treatment. Medication was allocated in a double-blinded manner and was to be taken by mouth before breakfast. Investigators at each research centre were responsible for patient enrolment and implementation of the random allocation sequence.
The primary efficacy endpoint of this study was the proportion of responders, defined as patients who had an average of three or more SCBM/week over 12 weeks of treatment. Previous studies conducted by the sponsor suggested a response rate of approximately 15% for placebo and 30% for prucalopride 2 mg. Taking into account two between-treatment comparisons (prucalopride 2 mg and 4 mg vs placebo), 188 patients per treatment group were needed to detect a significant difference in response rates of this magnitude, based on 90% power and 2.5% two-sided type 1 error. It was assumed that 5% of patients would have insufficient diary data after random assignment to allow evaluation of their response over the entire 12-week, double-blind phase. Therefore, 198 randomly assigned patients were required per treatment group, or a total of 594 randomly assigned patients.
The trial was conducted in accordance with the ICH Good Clinical Practice Guidelines, the Declaration of Helsinki and local laws and regulations. The clinical trial protocol was reviewed and approved by the appropriate independent ethics committees. Participants gave written informed consent.
Eligibility
Adult patients (⩾18 years) of both genders with a history of chronic constipation, defined as having two or fewer SCBM/week for a minimum of 6 months before the selection visit, were eligible to enter the study. Patients also had to show one or more of the following symptoms for at least one-quarter of the time: lumpy/hard stools, a sensation of incomplete evacuation, or straining during defecation. Patients were asked to record their bowel habits in daily diaries during the 2-week, drug-free run-in phase to confirm the presence of their constipation. Patients were excluded if the constipation was drug-induced or secondary to endocrine, metabolic or neurological disorders, surgery, known or suspected organic disorders of the large intestine, or megacolon. Other exclusion criteria were uncontrolled cardiovascular, liver and lung diseases, impaired renal function (serum creatinine >180 μmol/l) and clinically significant abnormal laboratory values.
Disallowed medication
Laxatives were not allowed. However, patients who did not have a bowel movement for three or more consecutive days throughout the trial were allowed to take bisacodyl (Dulcolax) as a rescue medication. A maximum single dose of 15 mg (three tablets of 5 mg) of bisacodyl was prescribed. If this standard bisacodyl dose was insufficient, a dose increase was allowed, but only after the patient contacted the investigator. If no bowel movements were passed after an increase in the bisacodyl dose, an enema could be administered. Their use had to be documented in the patient’s diary. Rescue medications were not allowed within 48 h before or after the start of double-blind treatment.
Assessments
Bowel habit assessments
Patients recorded the date and time of drug intake, bowel movement characteristics (date and time, stool consistency, degree of straining during defecation and sensation of complete evacuation) and the date and time of the use of rescue medication in daily diaries from the screening visit to the end of treatment. The primary and secondary endpoints were derived from these diaries.
The primary efficacy endpoint was the proportion of patients having on average three or more SCBM/week. This was evaluated over the first 4 weeks of treatment and over the entire 12 weeks of therapy.
The main secondary efficacy endpoint was the percentage of patients with an average increase of one or more SCBM/week from baseline. Other endpoints were: the average number of SCBM/week; the percentage of bowel movements with normal consistency, no straining, or a sensation of complete evacuation; the median time to onset of first SCBM after intake of the first dose of trial medication and the average number of bisacodyl tablets or enemas used per week.
Severity of constipation, efficacy of treatment and symptom assessments
Patients rated the severity of constipation and efficacy of treatment using the patient global assessment questionnaire.27 In addition, symptom-related data were obtained from the patient assessment of constipation symptoms (PAC-SYM) questionnaire28 at baseline, 2, 4, 8 and 12 weeks. The 12 constipation-related symptoms were grouped into three subscales for stool, abdominal and rectal symptoms.
QOL assessments
Patients rated their health-related QOL at baseline, week 4 and week 12 using the validated patient assessment of constipation quality of life (PAC-QOL) questionnaire.29 Patients were asked to score 28 items relating to the effects of constipation on their daily lives. These items were grouped into four subscales: physical discomfort, psychosocial discomfort, worries and concerns and patient satisfaction. The patient satisfaction subscale (five items) was considered as the most important endpoint derived from the PAC-QOL questionnaire, describing satisfaction with treatment and bowel habit.
Safety assessments
Adverse events were reported at weeks 2, 4, 8 and 12. Vital signs were evaluated at screening, baseline and weeks 2, 4, 8 and 12. ECG, clinical laboratory tests and physical examinations were reported at screening, week 4 and week 12.
Statistical analyses
All efficacy analyses were performed on data from the intent-to-treat (ITT) population. The ITT population comprised all patients taking at least one dose of double-blind study medication and who provided any post-baseline data for one or more key efficacy variables. All patients who took at least one dose of study medication were included in the safety analysis. The Cochran–Mantel–Haenszel test controlling for differences between countries was used to compare treatment groups in the percentage of patients reaching three or more SCBM/week. Other nominal data such as the key secondary parameter were analysed in the same way. Holm’s procedure was used to correct for the multiple pairwise comparisons. A 5% level of significance was used. Patients who dropped out early (<14 days) or who collected insufficient diary information for a proper evaluation were considered as non-responders for the primary endpoint. Bowel movements were only considered spontaneous if they occurred more than 24 h after a last laxative intake.
RESULTS
Patient characteristics
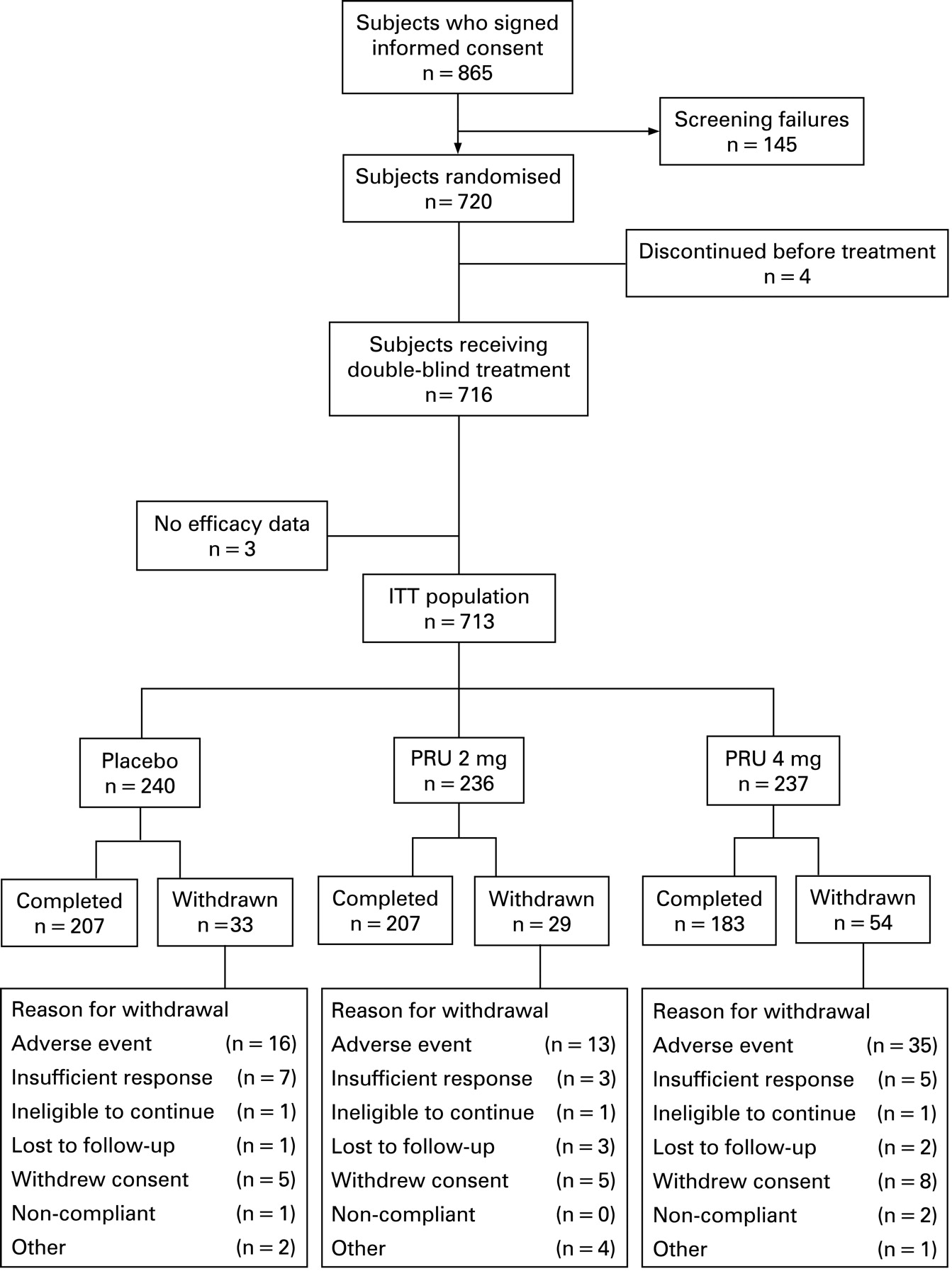
A total of 720 patients was randomly assigned to three treatment arms (prucalopride 2 mg, prucalopride 4 mg and placebo). Four patients discontinued before treatment start (fig 1) and three patients did not record any efficacy data and were all excluded from the ITT population (n = 713). Demographic data for the all (treated) patient population (n = 716) are shown in table 1. The mean duration of constipation was 17.5 years and 38.5% of patients reported no spontaneous bowel movements during the 6 months before study entry. Previous therapies for constipation were considered inadequate by 83.2% of patients. There were no significant differences in baseline characteristics between the treatment groups.

Primary efficacy endpoint
During the run-in, the average number of SCBM/week was 0.4–0.5 in each treatment group and a total of 58% of the patients had no SCBM. Prucalopride 2 mg or 4 mg treatment resulted in a significantly higher proportion of patients reaching an average of three or more SCBM/week over 12 weeks compared with placebo, reflecting normalisation of bowel function. Over 12 weeks, these proportions were 19.5% for 2 mg prucalopride (p⩽0.01 vs placebo), 23.6% for 4 mg prucalopride (p⩽0.001) and 9.6% for placebo (table 2). Over the first 4 weeks, an average of three or more SCBM/week was achieved by 23.7% on 2 mg prucalopride, 26.6% on 4 mg prucalopride and 10.4% on placebo (fig 2A) (p⩽0.001). This represents a therapeutic gain of approximately 10–15% with prucalopride over placebo. The superior efficacy of 2 mg prucalopride (p⩽0.01) and 4 mg prucalopride (p⩽0.001) compared with placebo in the proportion of patients with three or more SCBM/week was also observed during weeks 5–8 and 9–12 (fig 2A).

{kind=link}
{kind=link}
The subgroup of patients (83%) who reported dissatisfaction with previous (up to 6 months before the trial) laxative treatment showed similar response rates as the ITT population (data not shown).
Key secondary efficacy endpoint
The percentage of patients with an average increase of one or more SCBM/week over 12 weeks was 38.1% for 2 mg prucalopride, 44.1% for 4 mg prucalopride and 20.9% for placebo (p⩽0.001) (table 2). A subgroup analysis of 83% of patients who reported dissatisfaction with previous laxative treatment showed similar response rates (data not shown).
Other secondary efficacy endpoints
Other secondary efficacy parameters were significantly improved with both doses of prucalopride compared with placebo over 12 weeks (table 2). The median time to the first SCBM in both prucalopride groups was significantly shorter than in the placebo group (table 2). The mean decrease in the number of days with laxative use per week observed during the 12-week treatment period in both prucalopride groups was significantly greater than the change in the placebo group (p⩽0.001) (table 2). The prucalopride groups rated their constipation as less severe and were more satisfied with their treatment than the placebo group at week 12 (table 2).
Compared with placebo at week 12, prucalopride 2 mg or 4 mg also resulted in a significantly greater mean change in overall PAC-SYM scores (p⩽0.001) (table 3). Both doses of prucalopride also showed significantly greater improvements in the average change of scores on stool symptoms (p⩽0.001 both vs placebo) and on abdominal and rectal symptoms (p⩽0.05 for 2 mg and p⩽0.001 for 4 mg vs placebo) compared with placebo (table 3).
Primary QOL endpoint
On a scale of 0 to 4, with 4 representing the lowest satisfaction, the mean baseline scores for satisfaction ranged from 3.08 to 3.17 across the treatment groups, indicating a consistent and high level of dissatisfaction with treatment and bowel habit.
At week 12, the mean satisfaction subscale scores improved more in the prucalopride groups than in the placebo group. The average magnitude of improvement from baseline was two to three times greater in the prucalopride 2 mg and 4 mg groups (−0.76 and −0.87, respectively) than in the placebo group (−0.30; p⩽0.001) (table 4).
At week 12, the proportion of patients with an improvement of at least 1 point on the satisfaction subscale was significantly higher in the prucalopride 2 mg and 4 mg groups (33.5% and 29.4%, respectively) than in the placebo group (16.4%, p⩽0.001) (fig 2B).
Secondary QOL endpoints
The PAC-QOL overall score and other subscale scores (physical discomfort and worries and concerns subscale scores) were also significantly improved in both prucalopride groups compared with the placebo group at week 12 (table 4).
Safety
Treatment-emergent adverse events were reported by 170 (71.4%) patients on 2 mg prucalopride, 178 (74.8%) on 4 mg prucalopride and 161 (67.1%) on placebo (table 5). There were no deaths during the trial.
Overall, most adverse events were reported as mild or moderate in severity and were transient. The most frequently reported adverse events were headache, nausea, abdominal pain and diarrhoea. These adverse events were more prevalent in the prucalopride groups than in the placebo group (table 5), mainly on day 1 of treatment. Excluding the events occurring on day 1, the incidence of these adverse events was similar in the three treatment groups (table 5). The incidence of severe diarrhoea (based on the investigator’s assessment) was not higher in the prucalopride 2 mg and 4 mg groups (0.8% and 3.4%, respectively) compared with placebo (2.1%). There were no complaints of dehydration in 15 patients with severe diarrhoea; only two patients required antidiarrhoeal treatment and moderate hypokalaemia was reported in one patient on placebo.
Permanent discontinuation of trial medication as a result of adverse events was reported in 15 (6.3%) patients on 2 mg prucalopride, 36 (15.1%) on 4 mg prucalopride and 16 (6.7%) on placebo. The predominant adverse events leading to treatment discontinuation were headache, abdominal pain, nausea, diarrhoea and vomiting, often occurring in the first days of treatment. Discontinuation as a result of these adverse events accounted for the higher dropout rate in the prucalopride 4 mg group compared with other treatment groups.
There were no clinically relevant differences in haematology, clinical chemistry, urinalysis, vital signs, or ECG parameters (table 6) between the treatment groups. The overall incidence of post-baseline prolonged QT intervals (QTcF or QTcB) was low and was similar among all treatment groups.
DISCUSSION
In evaluating the overall treatment effect of prucalopride, it is important to take into account that enrolled patients had on average more than 15 years of constipation, a high degree of dissatisfaction with their bowel habits, a low QOL, a total of 58% of the patients had no SCBM/week during the run-in and more than 80% did not obtain adequate relief with previous laxative therapy. Therefore, the patient population analysed in this study represents a subgroup of patients that is significantly affected by their disease.
The primary endpoint in this trial, SCBM, was carefully chosen as a rigorous and clinically meaningful measure to evaluate the efficacy of prucalopride in a clinical trial. Compared with Rome III criteria that define constipation based partly on the frequency of bowel movements per week,30 SCBM adds a subjective measure of the completeness of evacuation to the objective measure of the number of bowel movements. Therefore, SCBM identifies bowel movements that fully relieve symptoms caused by chronic constipation.
Both doses of prucalopride resulted in a significantly higher proportion of patients achieving the primary efficacy endpoint, an average of three or more SCBM/week, compared with placebo. This frequency is commonly accepted as normal bowel habit. Overall, prucalopride provided 10–15% therapeutic benefit over placebo with this stringent endpoint. Recent clinical trial guidelines have suggested that a 10–15% improvement over placebo could be considered a clinically significant therapeutic gain in functional gastrointestinal disorders such as irritable bowel syndrome.31
Prucalopride treatment also showed significantly higher and clinically relevant increases than placebo in the proportion of patients with an average improvement of one or more SCBM/week from baseline. Similar response rates in primary and key secondary efficacy endpoints were seen in a large subgroup of patients who considered previous constipation therapies to be inadequate. Therefore, a large subgroup of patients with inadequate response to previous constipation therapies can be effectively treated with prucalopride. Both doses of prucalopride also improved other secondary efficacy endpoints, including stool consistency, evacuation completeness and constipation symptoms as measured by PAC-SYM. The improvement in the need for straining was significant in the 4 mg prucalopride group compared with placebo.
Laxatives have been shown to provide an increase of one and a half to two stools per week after 4 weeks of therapy, but evidence of their long-term efficacy is lacking.6–8 Many patients with chronic constipation are not satisfied with laxative treatment, primarily due to lack of efficacy.9 10 Patients taking laxatives report inadequate relief of bloating and other constipation symptoms, poor predictability of laxative treatment and lack of improvement in their QOL.9
Previous studies have shown that general wellbeing and QOL are lower among constipated patients than in healthy controls.4 32–34 Moreover, QOL decreases with the increasing severity of constipation.32–35
In this study, prucalopride not only increased bowel frequency and reduced constipation-related symptoms, but also improved patients’ perceptions of their disease and treatment. More patients treated with prucalopride compared with placebo scored their constipation as less severe and their treatment as quite or extremely effective. Prucalopride significantly improved patients’ satisfaction with bowel function and treatment as measured by PAC-QOL, the primary QOL endpoint. In addition, prucalopride improved assessments of the physical discomfort, psychosocial discomfort and worries/concerns associated with chronic constipation. Mean overall PAC-QOL scores were also significantly higher in the prucalopride groups compared with placebo after 12 weeks of treatment. Prucalopride thus improved health-related QOL in severely and chronically constipated patients. Similarly, a recent phase III study also showed that prucalopride significantly improved the severity of symptoms and health-related QOL in severely and chronically constipated patients compared with placebo.26
Our results also underscore the close relationship between patient satisfaction, QOL and the primary efficacy endpoint. Effective treatment of constipation and relief of symptoms were associated with significant increases in patient satisfaction and overall QOL. A 1-point improvement on the PAC-QOL scale is considered a high level of therapeutic responsiveness.29 Therefore, the improvements in patient satisfaction and overall QOL achieved with prucalopride can be considered clinically meaningful outcomes.26
In this trial, prucalopride was safe and well tolerated. The incidence of adverse events was comparable among the treatment groups, except for headache, nausea, vomiting and some gastrointestinal effects that were probably related to the pharmacological action of prucalopride. Previous motility agents targeting the 5-HT4 receptor, such as tegaserod and cisapride, have been associated with an unfavourable risk/benefit profile, potentially related to their interaction with 5-HT1B/D receptors and the hERG cardiac potassium channel, respectively.19 20 36–38 We observed no clinically relevant changes in vital signs or ECG parameters in patients taking prucalopride. The overall incidence of prolonged QT intervals was very low and was similar between the placebo and prucalopride groups. A review of the clinical safety database, which includes a total exposure of 406 patient-years in a double-blind, placebo controlled setting, identified no serious cardiovascular safety issues at doses of up to 20 mg/kg. Taken together with earlier clinical trials evaluating cardiovascular events24–26 39 40 and with in-vitro studies of the hERG channel,41 these data suggest a favourable cardiovascular profile for prucalopride.
CONCLUSION
Clinical evidence for the efficacy of laxative therapies is limited and generally does not support their long-term use in providing relief of symptoms.2 6–8 There remains a need for more effective and well-tolerated drugs to treat severe chronic constipation. Prucalopride 2 mg or 4 mg was effective, well tolerated and not associated with a higher incidence of cardiovascular complications compared with placebo in this study of patients with severe chronic constipation, the majority (83%) of whom were not adequately treated with laxatives. Prucalopride increased the number of SCBM/week, improved the completeness of bowel movements and reduced constipation symptoms and this efficacy was maintained throughout the 12-week treatment period. Prucalopride also improved patients’ satisfaction with bowel function and treatment, perceived severity of symptoms and QOL. Prucalopride 4 mg did not provide a meaningful benefit over the 2 mg dose. As there were fewer adverse events in the group treated with 2 mg compared with 4 mg prucalopride, the results currently support 2 mg prucalopride once daily for chronic constipation as the lowest effective dose. Further studies will be beneficial to determine the long-term efficacy and safety of prucalopride in larger populations as a constipation-reducing medication in the long term.
Acknowledgments
The authors acknowledge all site investigators for their participation in the study: R Batey, I Cook, F Dudley, T Florin, N Hoffman, J Kellow, N Yeomans (Australia); M Ferrante, F Huble, L Lepoutre, P van Eeghem, M van Outryve (Belgium); R J Bailey, C Bernstein, M Boivin, S Collins, G Devroede, R Fedorak, M Luterman, P Paré, W Paterson, R Reynolds, G K Turnbull (Canada); N Ahluwalia, M Benson, B Cooper, G Duthie, O Epstein, P Moayyedi, F Murray, C Pennington, C Smith, R Spiller, P Whorwell (UK); L M A Akkermans, G E Boeckxstaens, J G S Breed, R J F Felt-Bersma, L A Noach, M H Otten, C Sloots, T G Tan, M van Haastert, J H van Maanen, W J R R Venekamp, A M H Wetzels (The Netherlands); A Berstad, E Husebye, N Stray, H Waldum (Norway); D De Vos, D H. Grundling, K Pettengell, S Schmidt, H Schneider, C van Rensburg, T Winter, J Wright, J Zaidy (South Africa); T Hallgren, A Hugander, T Kjellin, I Magnussen, L Påhlman, S Skullman, H Tanghöj, C Tysk (Sweden).
REFERENCES
Footnotes
-
Funding: The study design and data collection were funded by Janssen Research Foundation, Beerse, Belgium. The analysis and interpretation of the data were funded by Movetis NV, Turnhout, Belgium.
-
Competing interests: JT has been a scientific advisor for Johnson & Johnson and Movetis. MvO has received consultancy fees from Movetis. GB, RK and LV are employees of Movetis NV, Turnhout, Belgium. Writing assistance was provided by Archimed Medical Communication Ag, Zofingen, Switzerland.
-
Ethics approval: The clinical trial protocol was reviewed and approved by the appropriate independent ethics committees.
-
Patient consent: Obtained.
Linked Articles
- Digest
- Miscellanea