Article Text

Abstract
Background We previously reported that microvesicles (MVs) released by human mesenchymal stem cells (MSC) were as effective as the cells themselves in both Escherichia coli lipopolysaccharide and live bacteria-induced acute lung injury (ALI) mice models. However, it remained unclear whether the biological effect of MSC MV can be applied to human ALI.
Methods In the current study, we tested the therapeutic effects of MSC MVs in a well-established ex vivo perfused human model of bacterial pneumonia. Using human donor lungs not used for transplantation, we instilled E. coli bacteria intrabronchially and, 1 hour later, administered MSC MVs into the perfusate as therapy.
Results After 6 hours, instillation of E. coli bacteria caused influx of inflammatory cells, which resulted in significant inflammation, lung protein permeability and pulmonary oedema formation. Administration of MSC MV significantly increased alveolar fluid clearance and reduced protein permeability and numerically lowered the bacterial load in the injured alveolus. The beneficial effect on bacterial killing was more pronounced with pretreatment of MSCs with a Toll-like receptor 3 agonist, polyinosinic:polycytidylic acid (Poly (I:C)), prior to the isolation of MVs. Isolated human alveolar macrophages had increased antimicrobial activity with MSC MV treatment in vitro as well. Although oxygenation and lung compliance levels were similar between injury and treatment groups, administration of MSC MVs numerically decreased median pulmonary artery pressure at 6 hours.
Conclusions In summary, MSC MVs increased alveolar fluid clearance and reduced lung protein permeability, and pretreatment with Poly (I:C) enhanced the antimicrobial activity of MVs in an ex vivo perfused human lung with severe bacteria pneumonia.
- ards
- pneumonia
- pulmonary oedema
Statistics from Altmetric.com
Key messages
What is the key question?
Can the therapeutic effects of mesenchymal stem cells (MSC) microvesicles, as reported in preclinical small animal models of acute lung injury, be applicable to human lung injury models?
What is the bottom line?
Human bone marrow-derived MSC microvesicles reduced multiple indices of acute lung injury in an ex vivo perfused human lung injured with severe bacterial pneumonia.
Why read on?
Based on promising results, further studies are warranted in a large animal model of pneumonia with or without sepsis to provide additional preclinical data for possible clinical trials.
Introduction
Acute respiratory distress syndrome due to bacterial pneumonia and/or sepsis remains a common cause of respiratory failure in critically ill intensive care unit patients with high mortality rates despite appropriate antibiotic use and supportive care.1 2 New therapeutic modalities are needed to improve morbidity and mortality. Multiple preclinical studies have demonstrated significant therapeutic potential with mesenchymal stem/stromal cells (MSC) for acute lung injury (ALI) despite limited engraftment rates of 1%~5%.3 4 5 The therapeutic potential of MSCs appeared to arise in part from the secretion of growth factors such as keratinocyte growth factor(KGF),6 anti-inflammatory products such as PGE2 or lipoxin A4,7 8 antipermeability factors such as angiopoietin-1 (Ang1),9 10 and antimicrobial products such as LL-37 or Lipocalin-2.11 12 Based on the significant potential of MSC, several phase I and II clinical trials focused on safety are underway (NCT02097641, NCT01902082, Clinicaltrials.gov).13 However, there remains some concerns with allogeneic stem cell therapy such as tumour formation and haemodynamic instability due to their physical properties.
Recently, stem cell-derived therapy, such as MSC conditioned media (CM) or extracellular vesicles, have generated considerable interest due to similar therapeutic properties without the inherent challenges of using live cells. Ionescu et al 14 demonstrated that MSC CM reduced lung inflammation and injury following lipopolysaccharide (LPS)-induced ALI in a mouse. Using LPS-induced ALI in an ex vivo perfused human lung, we also found that MSC CM reduced extravascular lung water, improved lung endothelial barrier permeability and restored alveolar fluid clearance (AFC) in part by KGF secretion by MSCs.6 More recently, we demonstrated that MSC-derived extracellular vesicles were as biologically active as MSCs in Escherichia coli LPS and bacteria-induced ALI in mice in part through the transfer of mRNA and microRNA to target cells, which resulted in improved resolution of pulmonary oedema and increased antimicrobial activity.15 16 We and other investigators also found that pretreatment of MSCs, such as with Poly (I:C),17 carbon monoxide,18 hypoxia19 and so on, may be a viable method to enhance the therapeutic properties of the cells and its released microvesicles (MVs).
Extracellular vesicles, which are released by most cell types, can be broadly classified into exosomes, MVs and apoptotic bodies based largely on their size and origin. Interestingly, extracellular vesicles contain mRNA, microRNA, protein, surface receptors and potentially organelles, which may be involved in their function.20–22 However, whether stem cell-derived therapy such as extracellular vesicles can be translated to large animal or even human ALI models is unknown. In the current study, we hypothesised that MSC MVs could be effective in reducing inflammation and total bacterial load in a severe pneumonia model in the ex vivo perfused human lung and that preconditioning MSCs with Poly (I:C) would enhance the antimicrobial properties of the released MVs.
Methods
Ex vivo perfused human lung and measurement of AFC
Either the right or left lung, from the Northern California Transplant Donor Network, was perfused slowly with DMEM without Phenol Red+5% bovine serum albumin until a cardiac output of 0.25 L/min was reached and then ventilated (Harvard Apparatus, tidal volume 300 mL, respiratory rate=10/min, fractional inspired oxygen (FiO2)=21%, and positive end-expiratory pressure (PEEP)=5 cmH2O) (figure 1).23 AFC rate was then measured in the control lung lobe by the change in protein concentration of a bronchoalveolar lavage (BAL) fluid over 30 min using the following equation: AFC (%/h)=2 x (1 -(Ci/Cf)) × 100 (Ci and Cf=protein concentration at 5 and 35 min, respectively). If 0<AFC<10%/h, 109 colony forming units (CFU) of E. coli bacteria was instilled into the lower lobe, and fresh human blood (100 mL) was added to the perfusate. One hour after injury, either 200 µL of MSC MV (1×), 400 µL of MSC MV (2×), 200 µL of Toll-like receptor 3 (TLR-3) agonist polyinosinic:polycytidylic acid (Poly (I:C)) pretreated MSC MV, or 200 µL of normal human lung fibroblasts (NHLF) MV as cellular control was administered intravenously in a random fashion. At 6 hours, the AFC rate of the injured lobe was measured.

Schematic of the ex vivo perfused human lung preparation. Marginal human lungs that were not used for lung transplantation were obtained from the Northern California Transplantation Donor Network. Using a large organ Harvard bioreactor, the pulmonary artery of the right or left lung was initially cannulated and gently perfused with a crystalloid solution containing 5% albumin at a rate up to 250 mL/min. Once rewarmed, the lung was ventilated using a tidal volume of 300 mL with 5 cmH20 of PEEP and a respiratory rate 10 breaths per minute in room air. AFC rate was first measured in the upper control lobe. If AFC>0 and <10 %/hour, 109 CFU E. coli bacteria was administered into the lower lung lobe, and 100 mL of fresh human blood was added to the perfusate. After 6 hours, AFC was measured in the lower lobe as the primary endpoint. In separate experiments and randomly, 200 or 400 µL MSC derived microvesicles (MVs), Poly (I:C) pretreated 200 µL MSC MVs or 200 µL NHLF-derived MVs was administered into the perfusate at 1 hour. AFC, alveolar fluid clearance; LLL, left lower lobe; LUL, left upper lobe; MSC, mesenchymal stem cell; NHLF, normal human lung fibroblasts; Poly (I:C), polyinosinic:polycytidylic acid; RML, right middle lobe; RUL, right upper lobe.
Isolation of MVs derived from human MSCs
MVs were isolated from the conditioned medium of human bone marrow-derived MSCs,24 obtained from a National Institutes of Health repository at Texas A&M Health Science Center, and NHLFs using ultracentrifugation. Briefly, MSCs or NHLFs were grown in a T75 flask until 90% confluency and then serum starved in alpha minimum essential medium (αMEM) or fibroblast basal medium (FBM) supplemented with 0.5% Bovine Albumin Fraction (MP Biomedical LLC). After 48 hours, the conditioned medium was collected and centrifuged at 3000 rpm for 20 min to remove cellular debris and then at 100 000 g (Beckman Coulter Optima L-100XP Ultracentrifuge) to isolate the MVs for 1 hour twice at 4°C. The final sediment was resuspended in PBS and stored in −80°C. Ten microlitres of the resuspension were equivalent to MVs released by 1 million MSCs or NHLFs over 48 hours.
TLR-3 priming of MSC
In order to switch MSC towards a more immunoregulatory phenotype, TLR-3 agonist, Poly (I:C) (1 µg/mL, Sigma Aldrich),was cultured with MSCs for 1 hour.17 The cells were then washed twice in PBS and serum starved over 48 hours prior to isolation of MVs.
MSC MV characterisation
MSC MVs were labelled with PKH26 (Sigma Aldrich) to separate out vesicles from debris or with antibodies for CD9-FITC, control IgG1 k-FITC, CD44-FITC or control IgG2b k-FITC (eBioscience and BD Biosciences) for flow cytometry. A BD FACSAria Fusion Special Order (SORP) cell sorter (BD Biosciences) with 100 nm nozzle and ND filter 1 was used. The threshold was set on the SSC 200. Data were analysed by Diva software (BD Biosciences). For fluorescence detection, we used a 586/15 band pass filter for PKH26 and 525/50 band pass filter for fluorescein isothiocyanate (FITC)-labelled antibodies. An unstained sample was used to detect autofluorescence and set the photomultiplier for all the channels. Standard silica beads (Apogee Mix for Flow Cytometer, Apogee Flow Systems), with a similar refractive index of vesicles, was used to gate the MSC MVs. MSC MVs were also characterised by scanning electron microscopy and by using NanoSight NS 300 (Malvern Instruments).
Measurement of lung protein permeability using Evans Blue
At time T5 hours, we injected 400 mg of Evans Blue (Sigma Aldrich) into the perfusate. At time T6.5 hours, 1 mL of perfusate was removed, and then 2 L of PBS was used to flush the lung. Several pieces (1–2 g) from both control and injured lung lobes were then collected and incubated with formamide for 3 days to release the dye. We measured the concentration of Evans blue dye from each sample to calculate lung protein permeability.
Isolation and bacterial killing effect of human alveolar macrophage
Human alveolar macrophages were isolated following BAL with warm normal saline and plated with RPMI 1640 media in a transwell plate (200 000 cell/well) after washing twice with HBSS solution and stimulated with 1 µg/mL LPS with or without 90 µL of MSC MV. The next day, the cells were cultured with opsonized 107 CFU of E. coli bacteria for 90 min. The CFU of E. coli in the medium was then counted.
Statistical analysis
Results were expressed as the mean±SD or median with 25%–75% percentile (IQR), as appropriate. Whether the samples were normally distributed was determined by D’Agostino & Pearson or Shapiro-Wilk normality test. Comparisons between several groups were made using the analysis of variance with Bonferroni correction if normally distributed or by Kruskal-Wallis test followed by post hoc Dunn’s tests if not. We used the software GraphPad Prism 7.0c.
Results
Baseline demographics and clinical data for donor human lungs
The demographic and clinical data for 37 donor lungs used are listed in the table 1. There appeared to be no major differences between the MV treatment groups compared with E. coli injury group in terms of age, sex, partial pressure of oxygen (PaO2)/FiO2 ratio, lung ischaemic time and lung injury score.
Clinical data of donor lungs
MSC MV characterisation
Scanning electron microscopy showed that the isolation technique yielded homogeneous population of spheroid particles (online supplemental figure 1). NanoSight analyses showed MSC MV mean size was 180±14 nm with a concentration at 4.7±0.2×108 particles per mL. For flow cytometry, we labelled MSC MV with PKH26 to quantify only membrane bound vesicles and to exclude debris. As a percentage of PKH26-labelled MSC MV, we found that 43%±8% was CD44 positive. An IgG Ab was used as a control to remove background staining. For CD9, only 0.1%±0.1% of PKH26 MSC MVs was labelled positively, suggesting that the majority of vesicles were MVs. However, since there is significant overlap between exosomes and MVs in terms of size, it is possible that the total number of exosome were under-represented. The MSC MV dose chosen was initially based on prior studies using MSCs in the ex vivo perfused human lung and our observation that MVs were approximately 5–-10× less potent than live cells.23 Consequently, we administered 200 µL of MSC MVs or the MVs released by 20 million MSCs over 48 hours; previous studies using the ex vivo human lung used 5–10 million MSCs as a therapeutic.
Supplementary file 1
MSC MVs improved AFC in lungs injured following E. coli pneumonia
Administration of either human MSC MVs (1× or 2×) or the Poly (I:C) MSC MVs into the perfusate at 1 hour following E. coli-induced ALI significantly increased AFC at 6 hours by 247% (p<0.001), 159% (p=0.001) or 180% (p<0.001), respectively. The AFC values were also significantly elevated compared with the control lung lobe by 144% (p<0.001), 82% (p=0.008) or 97% (p<0.001), respectively, suggesting a restoration and an improvement. In contrast, administration of NHLF MVs had no benefit and actually significantly decreased AFC compared with the control group (p=0.002) (figure 2A). Surprisingly, there was no dose effect with increasing MSC MV by 2× on any parameter, suggesting that there was a therapeutic ceiling to the MVs. By histology, instillation of intrabronchial E. coli bacteria caused significant injury as depicted by an increase in cellularity and blood, oedema and interstitial thickening, which was substantially reduced with the administration of either MSC MV (1× or 2×) or Poly (I:C) MSC MVs (figure 2B).

Therapeutic effect of MSC MVs on alveolar fluid clearance rate (AFC) following severe Escherichia coli pneumonia in ex vivo perfused human lung. (A) Administration of MSC MVs or Poly (I:C) pretreated MSC MVs intravenously significantly increased the AFC rate in the lung lobe injured by E. coli pneumonia at 6 hours. Administration of NHLF MVs had no beneficial effect on AFC rate. AFC was measured by the change in protein concentration of a 5% albumin instillate in the lung lobe over 1 hour and expressed as mean AFC (% per h, per 150 mL of BAL fluid) with 95% CIs for each condition. n=9 for E. coli-injured lung lobe, n=6 for E. coli-injured lung lobe treated with MSC MVs (200 µL or 400 µL) or n=11 for 200 µL MSC MVs pretreated Poly (I:C) or n=5 for 200 µL NHLF MVs. Overall p value <0.001; *p is significant versus control, **p is significant versus E. coli injured and ***p is significant versus NHLF MVs by analysis of variance (Bonferroni). P values and CIs for individual pair comparisons are shown in supplementary table. (B) Human lungs injured with E. coli bacteria with and without MSC MVs 1× or Poly (I:C) pretreated MSC MVs were fixed in 10% formalin at 6 hours. Sections were stained with H&E. Administration of human MSC MVs 1× or Poly (I:C) pretreated MSC MVs 1 hour after E. coli pneumonia injury reduced the level of haemorrhage, oedema and cellularity in the injured lung lobe at 6 hours following E. coli pneumonia. MSC,mesenchymal stem cell; MVs, microvesicles; NHLF, normal human lung fibroblasts; Poly (I:C), polyinosinic:polycytidylic acid.
Supplementary file 2
Poly (I:C) MSC MVs decreased bacterial count in the injured alveolus
Bacterial CFUs were measured in the BAL fluid following instillation of 109 E. coli bacteria with or without MV treatment at 6 hours. Although the median values of CFU levels in the MSC MV 1× and 2× treatment groups were numerically lower than the E. coli injury group, only the administration of Poly (I:C) MSC MVs significantly decreased CFU counts compared with the E. coli injured group at 6 hours by post hoc Dunn’s analyses following Kruskal-Wallis test (p=0.007). (figure 3A). Administration of NHLF MVs had no significant effect. The bacterial CFU counts in perfusate were also numerically lower in the MSC MV 2× and Poly (I:C) MSC MV groups compared with E. coli group at 6 hours but not statistically significant (figure 3B).

Therapeutic effect of MSC MVs on bacterial levels following severe Escherichia coli pneumonia in ex vivo perfused human lung. Administration of Poly (I:C) pretreated MSC MVs decreased (A) bacterial counts in the BAL fluid at 6 hours. Administration of NHLF MVs had no beneficial effect. Data are expressed in the box plot as median bacterial CFU counts with IQR for bars in the BAL fluid. Overall p value=0.016; **p is significant versus E. coli injury group by post hoc Dunn’s analyses following Kruskal-Wallis Test. (B) Although not statistically significant, administration of MSC MVs or Poly (I:C) pretreated MSC MVs numerically decreased bacterial CFU counts in the perfusate. Data are expressed in the graph as median bacterial CFU counts with IQR for bars in the perfusate. Overall p value =0.624. (C) Coincubation of MSC MVs with freshly isolated human alveolar macrophages also increased E. coli bacterial phagocytosis by the macrophages. Data are expressed in the graph as median bacterial CFU counts (as % of LPS control) with IQR for bars. Overall p value=0.026; *p is significant versus LPS-treated group by post hoc Dunn’s analyses following Kruskal-Wallis test. P values and CIs for individual pair comparisons are shown in supplementary table. CFU, colony forming units; LPS, lipopolysaccharide; MSC, mesenchymal stem cell; MVs, microvesicles; NHLF, normal human lung fibroblasts.
When freshly isolated human alveolar macrophages were cocultured with E. coli bacteria with or without LPS and/or MSC MVs, bacterial phagocytosis by alveolar macrophages significantly increased with MSC MV treatment (p=0.044) (figure 3C).
MSC MVs decreased absolute neutrophil count in the injured alveolus
Absolute neutrophil counts in the BAL fluid following E. coli pneumonia were numerically lower following administration of MSC MV (1×) or Poly (I:C) MSC MV at 6 hours (median 9.1×106 with IQR of 8.6×106–2.6×107 for E. coli, median 5.5×106 with IQR of 3.9×106–9.1×106 for +MSC MV 1X, median 7.2×106 with IQR of 4.7×106–9.9×106 for +Poly (I:C) MSC MV, n=6–11), with overall p value of 0.024 (figure 4A). However, no pair comparisons showed statistical significance after Dunn’s correction. There were also no significant differences in absolute neutrophil counts in the perfusate (data not shown).

Effect of MSC MVs on the influx of inflammatory cells following severe Escherichia coli pneumonia in ex vivo perfused human lung. Instillation of MSC MVs numerically decreased the influx of inflammatory cells, specifically neutrophils, into the lung lobe injured by E. coli pneumonia at 6 hours. Absolute neutrophil count is expressed as median with IQR for bars for each condition, n=9 for E. coli injured lung lobe, n=6–11 for E. coli–injured lung lobe treated with 200 µL or 400 µL of MSC MVs or 200 µL of Poly (I:C) pretreated MSC MVs. Overall p value =0.024. P values and CIs for individual pair comparisons are shown in supplementary table. MSC, mesenchymal stem cell; MVs, microvesicles.
MSC MVs decreased lung protein permeability
Lung protein permeability was measured in all lung lobes using the extravasation of Evans Blue dye, similar to the technique used in mice (figure 5A). Administration of MSC MVs (1× or 2×) or Poly (I:C) MSC MVs significantly reduced lung protein permeability (p=0.001–0.003) (figure 5B).

Therapeutic effect of MSC MVs on lung protein permeability following severe Escherichia coli pneumonia in ex vivo perfused human lung. Lung protein permeability at 6 hours as measured by Evan’s Blue extravasation. (A) Administration of Evan’s Blue intravenously demonstrated albumin protein leak into the E. coli bacteria injured lung lobe. (B) Compared with E. coli bacteria injured lung lobe, administration of MSC MVs or Poly (I:C) pretreated MSC MVs significantly reduced lung protein permeability. Data are presented as mean change in permeability with 95% CIs. Overall p value=0.001. *P is significant versus E. coli by analysis of variance (Bonferroni). P values and CIs for individual pair comparisons are shown in supplementary table. MSC, mesenchymal stem cell; MVs, microvesicles; Poly (I:C), polyinosinic:polycytidylic acid.
Effects of MSC MVs on pulmonary arterial pressure (PAP) and pulmonary vascular resistance (PVR)
PAP was measured throughout the duration of the experiment. Although not statistically significant, administration of MSC MVs or Poly (I:C) MSC MVs lowered PAP at T6 hours compared with the E. coli injury group (median 3.6 with IQR of 0–18 for E. coli, median 0.2 with IQR of 0–3.3 for +MSC MV 1×, median 0 with IQR of 0–1.4 for +MSC MV 2×, median 0 with IQR of 0–1.1 for +Poly (I:C) MSC MV, n=6–11) (figure 6A). PVR followed a similar trend (figure 6B).
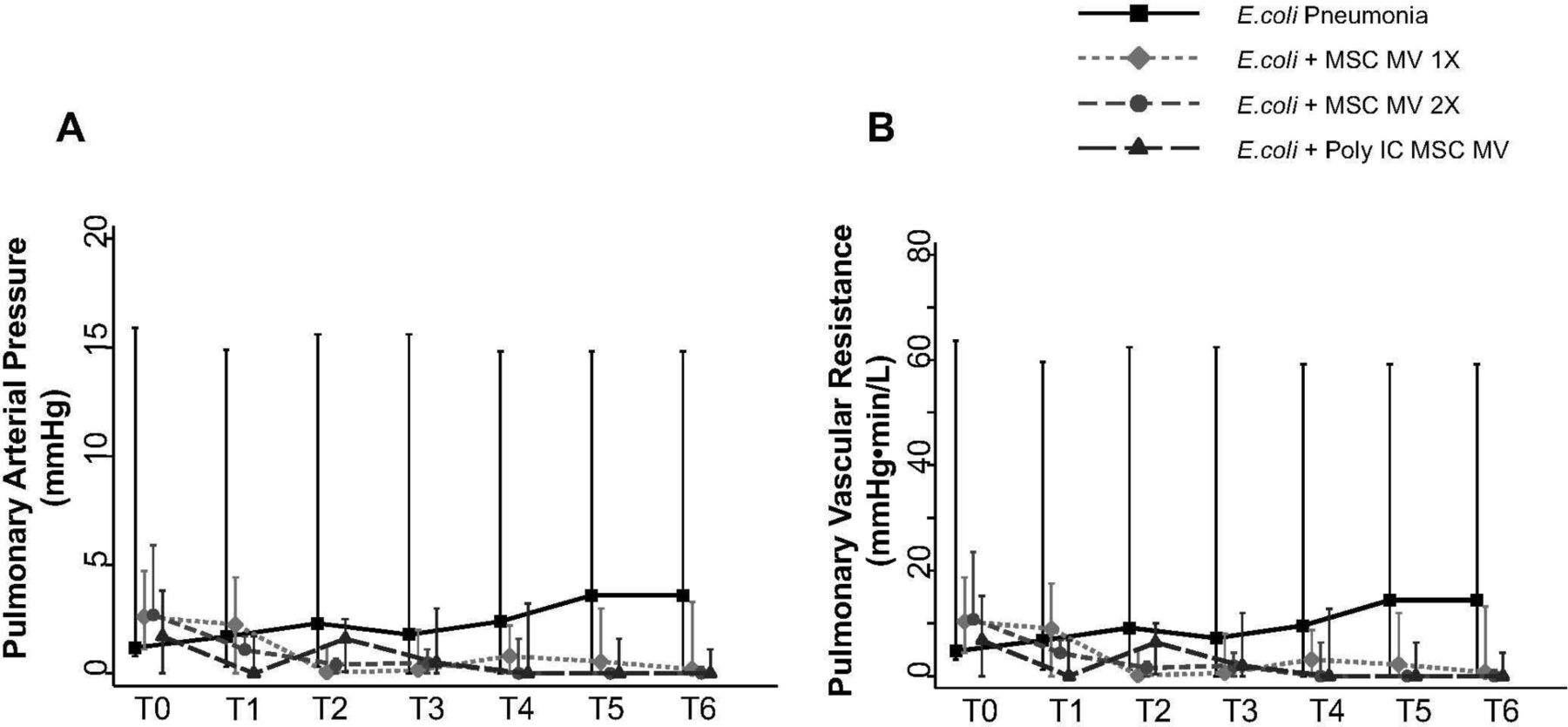
Effect of MSC MVs on pulmonary arterial pressure and vascular resistance following severe Escherichia coli pneumonia in ex vivo perfused human lung. Administration of MSC MVs or Poly (I:C) pretreated MSC MVs numerically decreased PAP (A) or PVR (B) at T6 hours compared with E. coli injury group. Data are presented as median PAP or PVR with IQR as bars. P=0.20–0.66 for each time point by Kruskal-Wallis tests (A). P=0.20–0.63 for each time point by Kruskal-Wallis tests (B). MSC, mesenchymal stem cell; MVs, microvesicles; PAP, pulmonary arterial pressure; Poly (I:C), polyinosinic:polycytidylic acid; PVR, pulmonary vascular resistance.
No effect of MSC MVs on tracheal pressure, lung compliance or oxygenation
There were no significant differences in tracheal pressure or lung compliance between control and treatment groups (figure 7A,B). There was also no change in PaO2 between groups (figure 7C), possibly due to the short duration of the experiment and the technique used to measure AFC.

{kind=link}
![[SP1.jpg]](https://thorax.bmj.com/content/thoraxjnl/74/1/43/DC1/embed/inline-supplementary-material-1.jpg?download=true){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lack of a therapeutic effect of MSC MVs on lung compliance or oxygenation following severe Escherichia coli pneumonia in ex vivo perfused human lung. Administration of MSC MVs or Poly (I:C) pretreated MSC MVs had no significant effect on tracheal pressure (A), lung compliance (B) or oxygenation (C) over 6 hours. Data are presented as median tracheal pressure or lung compliance or PaO2 with IQR as bars. P=0.56–0.98 for each time point by Kruskal-Wallis tests (A). P=0.56–0.98 for each time point by Kruskal-Wallis tests (B). P=0.10–0.92 for each time point by Kruskal-Wallis tests (C). MSC, mesenchymal stem cell; MVs, microvesicles; PaO2, arterial oxygen tension (or pressure); Poly (I:C), polyinosinic:polycytidylic acid.
Effect of MSC MV on tumour necrosis factor (TNF)α levels in the injured alveolus
We measured the inflammatory cytokine (TNFα) in the BAL fluid. Although not statistically significant, administration of human MSC MV 1× after E. coli pneumonia numerically reduced the levels of TNFα (pg/ml) by 72% (median 5285 with IQR of 3446–13 409 for E. coli, median 1454 with IQR of 681–8097 for +MSC MV 1×, median 5105 with IQR of 3263–9285 for +MSC MV 2X, median 6123 with IQR of 1766–9152 for +Poly (I:C) MSC MV, n=6–11).
Discussion
The main findings of these studies can be summarised as follows: (1) intravenous administration of MSC MVs significantly increased the rate of AFC and decreased lung protein permeability and numerically lowered the CFU bacteria counts and inflammation in the injured alveolus following severe E. coli pneumonia in an ex vivo perfused human lung (figures 2, 3 and 5); (2) Poly (I:C) pretreatment of MSCs significantly enhanced the antimicrobial property of the released MVs in the injured alveolus (figure 3); (3) increased antimicrobial effect of Poly (I:C) MSC MVs was associated with a numerically lower influx of inflammatory cells (ie, neutrophils) in the injured lung (figure 4); (4) MSC MVs and Poly (I:C) MSC MVs treatment groups had numerically lower median PAP and PVR levels at 6 hours (figure 6). However, MSC MVs and Poly (I:C) MSC MVs did not improve tracheal pressure, lung compliance or oxygenation (figure 7); (5) freshly isolated human alveolar macrophage had enhanced antimicrobial properties with MSC MV treatment (figure 3); and (6) more significantly, the preclinical therapeutic findings seen with MSC MVs in mice were apparent in a clinically relevant human lung injury model, especially in the effect of Poly (I:C) MSC MVs, making it a possible viable alternative to MSCs as a therapy.
Most cell types release MVs,25 which can be biologically active as mediators of cell-to-cell communication or through direct effects on target cells.26–28 Recently, MVs derived from MSC have been reported to exert therapeutic effects on various organ injury models.29 30 We previously found that MSC MV significantly reduced pulmonary oedema in mice injured with LPS or E. coli pneumonia15 16 and an ex vivo perfused human lung model of ischaemia/reperfusion.31 However, the underlying mechanisms were not fully known. In the current study, both MSC MVs and Poly (I:C) MSC MVs improved AFC to a near-normal level at 6 hours following E. coli pneumonia (figure 2). We previously demonstrated that MSC and/or MSC MVs increased AFC rate in part through enhanced KGF expression; KGF secreted from MSCs upregulated the key sodium channel (αENaC) in alveolar epithelial cells, increasing fluid absorption.6
We also found that Poly (I:C) MSC MVs enhanced bacterial killing in vivo and MSC MVs in vitro (figure 3) and numerically reduced the influx of neutrophils (figure 4) into the injured alveolus. Several potential mechanisms may account for these observations: (1) MSC and MSC MVs secrete or enhance the expression of antimicrobial soluble factors. Krasnodembskaya et al found that MSCs secreted LL-37, a known antimicrobial peptide, and Gupta et al 11 12 demonstrated that MSCs enhanced survival and bacterial clearance through upregulation of Lipocalin-2 by MSCs in a mice model of E. coli pneumonia; (2) Raffaghello et al 32 demonstrated that MSCs inhibited apoptosis of neutrophils by MSC through downregulation of reactive oxygen species and Stat-3 phosphorylation; and (3) multiple groups demonstrated that MSC and MSC MVs enhanced alveolar macrophage survival via mitochondria transfer (via tunnelling nanotubes) and phenotypic change from the proinflammatory M1 to the anti-inflammatory M2.33–35 Both Nemeth et al and Mei et al 7 36 found that MSCs upregulated macrophage phagocytic activity, which improved bacterial clearance in a caecal ligation and puncture model of sepsis. In addition, recent studies indicated that TLR-3 activation with Poly (I:C) can enhance the immunomodulatory potential of MSC, while TLR-4 activation could reduce it.17 In a mice model of E. coli pneumonia, we found that Poly (I:C) pretreatment of MSC MV enhanced macrophage/monocyte phenotypic M2 change, as indicated by decreased inflammation and increased bacteria phagocytosis.16
MSC MVs and Poly (I:C) MSC MVs both significantly reduced lung protein permeability (figure 5), which may be attributed to its Ang1 content, a protein with well-known anti-inflammatory, antipermeability and endothelial protective characteristics. Both MSCs and MSC MVs have significant levels of Ang1 mRNA. In LPS-induced ALI in mice, MSC or MSC transfected with the human Ang1 gene reduced pulmonary vascular injury and the recruitment of inflammatory cells into the lung.10 More recently, Tang et al 37 found that Ang1 in MSC MV was important in reducing inflammation and pulmonary oedema in LPS-induced ALI in mice.
Our study has some limitations: (1) lack of an intact lymphatic system for lung interstitial fluid clearance, which may be relevant if AFC is measured over longer periods; (2) lack of other immune organs such as the spleen or liver, which may participate in injury and/or repair; (3) short duration of lung injury, which limits assessment of whether the effects of MSC MVs can be sustained; and (4) limited sample size due to lack of availability of lungs for research.
Despite the limitations, the importance of the current study was in establishing the use of MVs as a possible viable alternative to using live cells in ALI. The benefits include ease of isolation and storage, avoiding dimethyl sulfoxide for preservation and the need for a bone marrow transplant facility, avoidance of using live stem cells with tumourigenic potential and which may cause haemodynamic instability with administration and potential to modify MVs with pretreatment of the cells to enhance the therapeutic effects. However, the major obstacle remains in scaling the production of MSC MVs since the potency is approximately 5–10× less than the cells. Given that a typical MSC dose is 5 million cells/kg or 500 million cells for a 100 kg patient, future clinical trials with MSC MVs may require isolating MVs released from up to 5 billion cells per patient, which may be prohibitive. Studies are ongoing to address this critical issue.
In conclusion, our study demonstrated that MSC MVs was therapeutic in a clinically relevant human model of severe E. coli pneumonia in lungs with baseline injury. Based on these promising results, further studies are warranted in a large animal with pneumonia and or sepsis to provide further biological rationale and additional preclinical data for possible clinical trials.
References
Footnotes
Contributors JP: conception and design, collection and assembly of data, data analysis and interpretation, and manuscript writing. SK, HL, AL, SH, JL, HZ and QH: collection and assembly of data and data analysis and interpretation. MAM: financial support, data analysis and interpretation and manuscript writing. J-WL: conception and design, financial support, data analysis and interpretation, manuscript writing and final approval.
Funding This work was supported by the National Institute of Health National Heart, Lung, and Blood Institute grant number HL-113022 (Dr J-W Lee), HL-51856 and HL-131621 (Dr M A Matthay).
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial
- Airwaves