Article Text

Statistics from Altmetric.com
The term “cor pulmonale” is still very popular in the medical literature, but its definition varies and there is presently no consensual definition. Forty years ago an expert committee of the World Health Organization1 defined cor pulmonale as “hypertrophy of the right ventricle resulting from diseases affecting the function and/or structure of the lungs . . .”. This pathological definition is in fact of limited value in clinical practice. It has been proposed to replace the term “hypertrophy” by “alteration in the structure and function of the right ventricle”. It has also been proposed to define clinically cor pulmonale by the presence of oedema in patients with respiratory failure. Finally, as pulmonary arterial hypertension is “the sine qua non” of cor pulmonale,2 we believe that the best definition of cor pulmonale is : pulmonary arterial hypertension resulting from diseases affecting the structure and/or the function of the lungs; pulmonary arterial hypertension results in right ventricular enlargement (hypertrophy and/or dilatation) and may lead with time to right heart failure.
A new diagnostic classification of pulmonary hypertension was developed by a group of experts in 19983 and is presented on table 1. In our opinion cor pulmonale corresponds to the third part of this classification (pulmonary hypertension associated with disorders of the respiratory system and/or hypoxaemia) and must be distinguished from pulmonary venous hypertension (part 2), and also from primary pulmonary hypertension (part 1) and from thromboembolic pulmonary hypertension (part 4).
New diagnostic classification of pulmonary hypertension3
This article reviews the current state of knowledge about pulmonary hypertension resulting from disorders of the respiratory system and/or from chronic hypoxaemia. We will only consider chronic cor pulmonale. Particular emphasis will be placed on chronic obstructive pulmonary disease (COPD) which is by far the main cause of cor pulmonale.
DEFINITIONS AND EPIDEMIOLOGY
Pulmonary hypertension complicating chronic respiratory disease is generally defined by the presence of a resting mean pulmonary artery pressure (PAP) > 20 mm Hg. This is slightly different from the definition of primary pulmonary hypertension (PAP > 25 mmHg).3 In young (< 50 years) healthy subjects PAP is most often between 10–15 mm Hg. With aging there is a slight increase in PAP, by about 1 mm Hg/10 years. A resting PAP > 20 mm Hg is always abnormal. In the “natural history” of COPD, pulmonary hypertension is often preceded by an abnormally large increase in PAP during exercise, defined by a pressure > 30 mm Hg for a mild level of steady state exercise. The term “exercising” pulmonary hypertension has been used by some authors, but the term “pulmonary hypertension” should be reserved for resting pulmonary hypertension.
Cor pulmonale is a common type of heart disease, as a result of its close association with COPD which has emerged, in recent years, as a leading cause of disability and death.4 But there are in fact very few data about the incidence and prevalence of cor pulmonale. The main reason is that right heart catheterisation cannot be performed on a large scale in patients at risk. An alternative approach is the use of non-invasive methods, particularly Doppler echocardiography. It should be possible to investigate large groups of respiratory patients with echo Doppler within the next few years.
A UK study performed in Sheffield5 has tried to determine the prevalence of patients at risk of developing pulmonary hypertension and cor pulmonale—that is, patients with hypoxaemic lung disease. In the study population, aged ≥ 45 years, an estimated 0.3% had both an arterial oxygen tension (Pao2) < 7.3 kPa (55 mm Hg) and a forced expiratory volume in one second (FEV1) < 50% of the predicted value. For England and Wales this could represent 60 000 subjects at risk of pulmonary hypertension and eligible for long term oxygen therapy.
The mortality related to cor pulmonale is also difficult to assess. There are data about the mortality resulting from chronic lung disease (100 000/year in the USA4) but we do not know precisely the role of secondary pulmonary hypertension in this mortality. Pulmonary hypertension is a complication, among others, of advanced COPD and it is not possible to separate it from its causative diseases.
AETIOLOGY: WHICH CHRONIC LUNG DISEASE MAY LEAD TO COR PULMONALE?
Table 2 lists the chronic respiratory diseases which may lead to cor pulmonale. Primary pulmonary hypertension, pulmonary thromboembolic disease, and diseases of the pulmonary vascular bed have been excluded from this list which is far from exhaustive. There are three major groups of diseases:
those characterised by a limitation to airflow (COPD and other causes of chronic bronchial obstruction)
those characterised by a restriction of pulmonary volumes from extrinsic or parenchymatous origin (restrictive lung diseases)
those where the relatively well preserved mechanical properties of the lungs and chest wall contrast with pronounced gas exchange abnormalities which are partially explained by poor ventilatory drive (respiratory insufficiency of “central” origin).
Diseases of the respiratory system associated with pulmonary hypertension (except primary pulmonary hypertension, pulmonary thromboembolic disease, and diseases of the pulmonary vascular bed)
COPD is the major cause of chronic respiratory insufficiency and cor pulmonale, and it probably accounts for 80–90% of the cases. COPD includes chronic obstructive bronchitis and emphysema which are often associated. Among the restrictive lung diseases kyphoscoliosis, idiopathic pulmonary fibrosis, and pneumoconiosis are the main causes of cor pulmonale. Among the aetiologies of respiratory insufficiency of “central” origin the obesity–hypoventilation syndrome (formerly “Pickwickian syndrome”) is a relatively frequent cause of cor pulmonale.
MECHANISMS OF COR PULMONALE
As stated above pulmonary hypertension is the “sine qua non” of cor pulmonale. Accordingly, the mechanisms of cor pulmonale are first those of pulmonary hypertension. In chronic respiratory diseases pulmonary hypertension results from increased pulmonary vascular resistance (PVR) whereas cardiac output and pulmonary “capillary” wedge pressure are normal; pulmonary hypertension is said to be precapillary.
The factors leading to an increased PVR in chronic respiratory disease are numerous but alveolar hypoxia is by far the most predominant,2 at least in COPD, kyphoscoliosis, and the obesity–hypoventilation syndrome. Two distinct mechanisms of action of alveolar hypoxia must be considered: acute hypoxia causes pulmonary vasoconstriction, and chronic longstanding hypoxia induces structural changes in the pulmonary vascular bed (pulmonary vascular remodelling).
Hypoxic pulmonary vasoconstriction (HPV) has been known since the studies in 1946 of Von Euler and Liljestrand on the cat. HPV explains the rise of PVR and PAP observed in humans, and in almost all species of mammals, during acute hypoxia. This vasoconstriction is localised in the small precapillary arteries. Its precise mechanism is not fully understood. The clinical situations which bear the closest analogy with acute hypoxic challenges are probably exacerbations of COPD leading to acute respiratory failure, and the sleep related episodes of worsening hypoxaemia.
Abbreviations
-
COPD: chronic obstructive pulmonary disease
-
FEV1: forced expiratory volume in one second
-
HPV: hypoxic pulmonary vasoconstriction
-
LTOT: long term oxygen therapy
-
MRI: magnetic resonance imaging
-
Pao2: arterial oxygen tension
-
Paco2: arterial carbon dioxide tension
-
PAP: pulmonary artery pressure
-
PVR: pulmonary vascular resistance
-
RHF: right heart failure
-
RVEF: right ventricular ejection fraction
Pulmonary hypertension is generally observed in respiratory patients exhibiting pronounced chronic hypoxaemia (Pao2 < 55–60 mm Hg). It is accepted that chronic alveolar hypoxia leads to remodelling of the pulmonary vascular bed (hypertrophy of the muscular media of the small pulmonary arteries, muscularisation of pulmonary arterioles, and intimal fibrosis) comparable to that observed in natives living at high altitude. This remodelling leads to elevation of PVR and to pulmonary hypertension. In fact the remodelling of the pulmonary vessels may be observed early in non-hypoxaemic COPD patients with mild disease severity.
Furthermore, other functional factors must be considered, namely hypercapnic acidosis and hyperviscosity caused by polycythaemia, but their role seems small when compared to that of alveolar hypoxia. In idiopathic pulmonary fibrosis the increase of PVR is caused by anatomical factors: loss of pulmonary vascular bed or compression of arterioles and capillaries by the fibrosing process.
Pulmonary hypertension increases the work of the right ventricle, which leads more or less rapidly to right ventricular enlargement (associating hypertrophy and dilatation) which can result in ventricular dysfunction (systolic, diastolic). Later, right heart failure (RHF) characterised by the presence of peripheral oedema can be observed, at least in some respiratory patients. The interval between the onset of pulmonary hypertension and the appearance of RHF is not known and may vary from one patient to another. There is a relation between the severity of pulmonary hypertension and the development of RHF.
CLINICAL ASSESSMENT OF COR PULMONALE: PLACE OF NON-INVASIVE METHODS
The clinical signs of cor pulmonale are relatively insensitive6 and some of them (signs related to an increased jugular venous pressure) are often obscured by hyperinflation of the chest6 which is present in a number of COPD patients. Furthermore, the clinical signs occur late, being observed at an advanced stage of the disease far after the development of pulmonary hypertension. Peripheral (ankle) oedema is the best sign of RHF but it is not specific and can arise from other causes; in some patients with pulmonary hypertension, it does not occur at all. A murmur of tricuspid regurgitation, suggesting right ventricular dilatation, is a very late sign in respiratory patients. Accentuation of the pulmonary component of the second heart sound is only observed in patients with severe pulmonary hypertension.
The detection of right ventricular hypertrophy by electrocardiography has a high specificity but a very low sensitivity. A normal ECG does not exclude the presence of pulmonary hypertension, particularly in COPD patients. Similarly, the radiological signs of pulmonary hypertension (increased width of the right descending pulmonary artery) are poorly sensitive and the radiological appearance of a dilated right ventricle is a very late (and inconsistent) sign.
The non-invasive diagnosis of pulmonary hypertension is presently based on echocardiography. Continuous wave Doppler echocardiography allows the calculation of the transtricuspid pressure gradient from the peak velocity of the tricuspid regurgitant jet, by applying the Bernouilli equation. Assuming a right atrial pressure of 5 mm Hg, it is thus possible to calculate right ventricular systolic pressure (right atrial pressure + transtricuspid pressure gradient) which is identical to pulmonary artery systolic pressure. It is also possible to estimate the diastolic pulmonary artery pressure by summing the right atrial pressure and the end diastolic pressure gradient between the pulmonary artery and the right ventricle. Pulsed wave Doppler echocardiography, also based on the measurement of flow velocity, allows an indirect estimation of pulmonary artery systolic pressure. However, hyperinflation makes echocardiography difficult in many COPD patients and a reliable examination cannot be obtained in more than 60–80% of the cases. The good correlations that have been observed in cardiac patients between PAP estimated from echo data and pressures measured invasively have not always been confirmed in COPD patients7 and a mean error of the estimate, for PAP, of about 10 mm Hg has been reported.7
Two dimensional echocardiography is used to measure right ventricular dimensions and the right ventricular wall thickness, making it possible to assess the presence of right ventricular hypertrophy and/or dilatation. However, magnetic resonance imaging (MRI) is probably the best method for measuring right ventricular dimensions because it produces the best images of the right ventricle. In COPD patients good correlations have been noted between right ventricular free wall volume measured by MRI and PAP. MRI is also a good method for detecting changes in right ventricular function, but it is expensive and available only in specialised centres.
Radionuclide ventriculography allows the measurement of right ventricular ejection fraction (RVEF). An RVEF < 40–45% is considered abnormal, but RVEF is not a good index of right ventricular function; it gives only an estimate of the systolic function and is afterload dependent, decreasing when PAP and PVR increase.8 Accordingly, the decreased RVEF observed in many COPD patients is caused primarily by increased afterload conditions and is not an indicator of “true” right ventricular dysfunction.
MAIN FEATURES OF PULMONARY HYPERTENSION IN CHRONIC RESPIRATORY DISEASE
The main characteristic of pulmonary hypertension in chronic respiratory disease is probably its mild to moderate degree of hypertension, with resting PAP in a stable state of the disease ranging usually between 20–35 mm Hg. This modest degree of pulmonary hypertension, well recognised in COPD,9 is very different from left heart disease, congenital heart disease, pulmonary thromboembolic disease, and particularly primary pulmonary hypertension, where PAP is usually > 40–50 mm Hg. Table 3 compares the pulmonary haemodynamic data of COPD patients9 with a large series of patients with primary pulmonary hypertension (US National Institutes of Health Registry). It can be seen that pulmonary hypertension is severe in primary pulmonary hypertension (mean (SD) PAP 60 (15) mm Hg) but is rather modest in COPD (PAP 26 (6) mm Hg). A PAP ≥ 40 mm Hg is unusual in COPD patients except when they are investigated during an acute exacerbation or when there is an associated cardiopulmonary disease. The consequences of this modest level of pulmonary hypertension include the absence or late occurrence of RHF and the frequent inability of non-invasive methods to achieve a diagnosis of pulmonary hypertension. However, pulmonary hypertension, even if mild at baseline, may worsen during exercise and sleep and during acute exacerbations of the disease.
Comparison of pulmonary hypertension in chronic hypoxic lung disease (COPD) to primary pulmonary hypertension
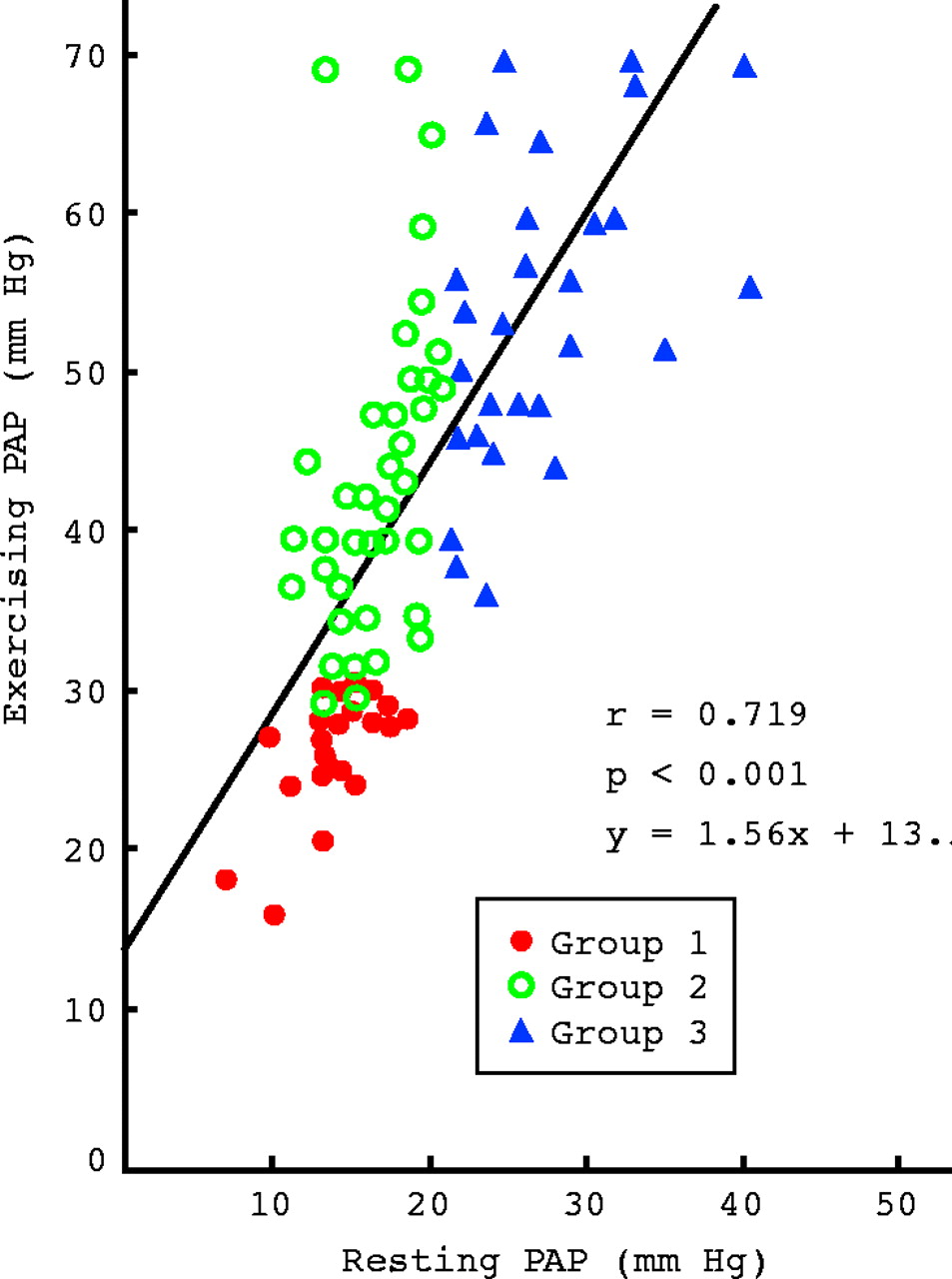
During steady state exercise PAP increases notably in advanced COPD patients with resting pulmonary hypertension,10 as illustrated in fig 1 which shows that in these patients (group 3) mean PAP rises from 27 mm Hg to 55 mm Hg during a 30–40 W exercise of 7–10 minutes duration. This is explained by the fact that PVR does not decrease during exercise in these patients; as the cardiac output is doubled for this level of exercise, PAP increases by about 100% (fig 1).
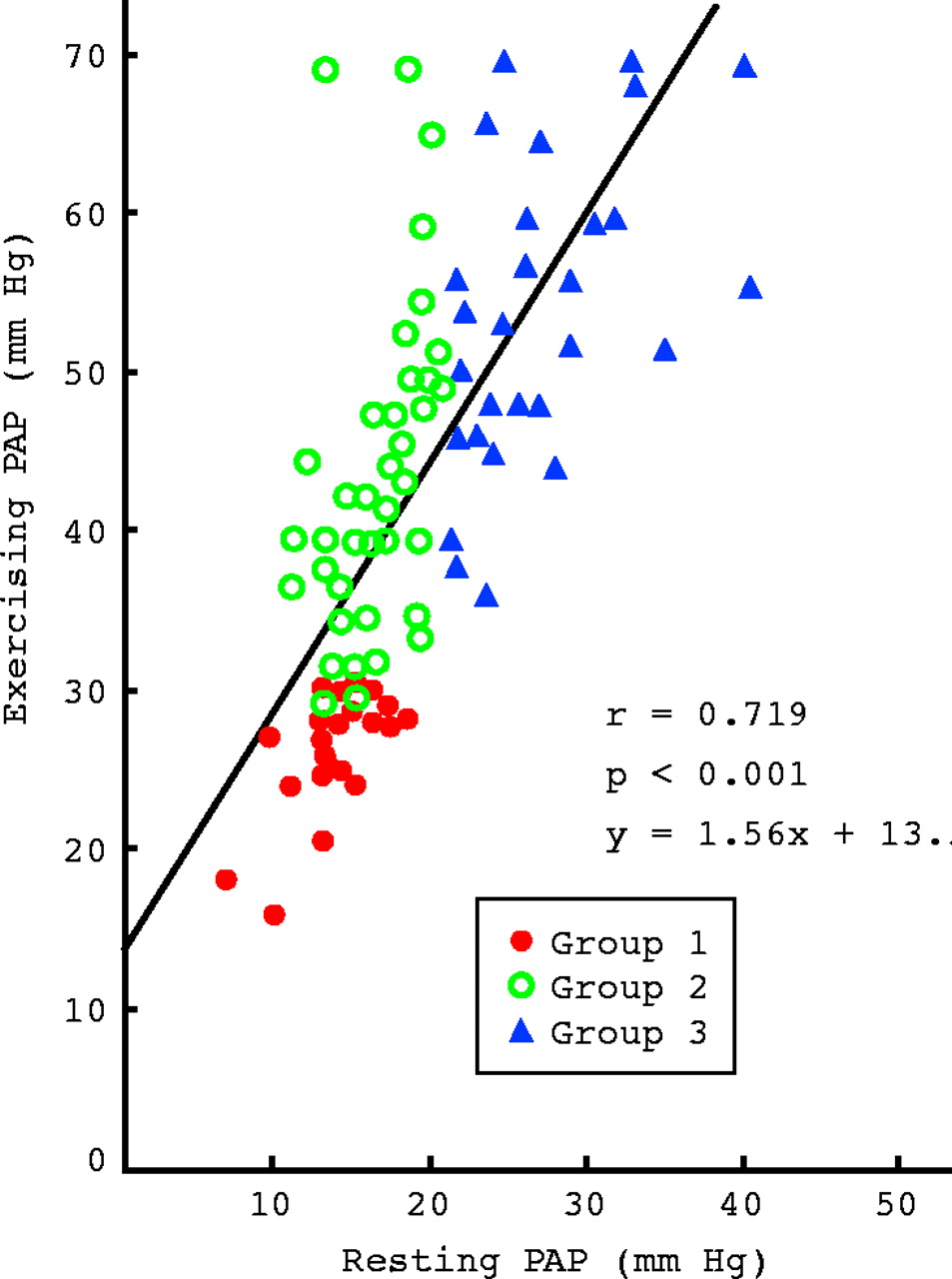
Resting and exercising pulmonary artery mean pressure (PAP) in a large series of chronic obstructive pulmonary disease patients (results are in mm Hg). Group 1: resting PAP < 20 mm Hg and exercising PAP < 30 mm Hg. Group 2: resting PAP < 20 mm Hg and exercising PAP > 30 mm Hg. Group 3: resting PAP > 20 mm Hg (pulmonary hypertension).
Acute increases of PAP during sleep have been observed in COPD patients with respiratory failure.11 They are principally observed in REM (rapid eye movement) sleep during which dips in oxygen saturation are more severe. These episodes of sleep related desaturation are not caused by apnoeas, except if COPD is associated with a sleep apnoea syndrome, but by alveolar hypoventilation and/or ventilation–perfusion mismatching.11 The more profound the dips of hypoxaemia, the more severe the peaks of pulmonary hypertension (PAP can increase by > 10 mm Hg from its baseline value).
Episodes of acute respiratory failure are characterised by a worsening of hypoxaemia and hypercapnia, and simultaneously there is a pronounced increase in PAP.12 PAP may increase by as much as 20 mm Hg but usually returns to its baseline after recovery, as shown in fig 2. The striking parallel between changes in Pao2 and PAP suggests the presence of HPV. The acute increases of afterload occurring during exercise, sleep, and exacerbations of the disease can favour the development of RHF.13

{kind=link}
{kind=link}
Evolution of arterial oxygen tension (Pao2) and mean pulmonary artery pressure (PAP) in a series of chronic obstructive pulmonary disease patients investigated during acute exacerbations and after recovery. The pronounced improvement in Pao2 (from mean 38 mm Hg to 53 mm Hg) is accompanied by a profound decrease in PAP (from mean 44 mm Hg to 27 mm Hg).
On the other hand the progression of pulmonary hypertension is slow, at least in COPD patients, and PAP may remain stable over periods of 3–10 years.14 In a series of 93 COPD patients followed up during a mean of 90 months, the average change in PAP was only +0.5 mm Hg/year for the group as a whole.14 This means that in the majority of COPD patients whose PAP is initially normal (< 20 mm Hg) it will not exceed 20 mm Hg after five years. But a minority (about 30%) of advanced COPD patients exhibit a notable worsening of PAP during the follow up14: these patients do not differ from the others at the onset, but they are characterised by a progressive deterioration of Pao2 and Paco2 during the evolution, which underlines the necessity for regular measurements of arterial blood gases for advanced COPD patients.
FROM PULMONARY HYPERTENSION TO RIGHT HEART FAILURE
The classical view of the development of RHF in chronic respiratory patients—that is, pulmonary hypertension leading to right ventricular enlargement, right ventricular dysfunction, and finally to RHF—has been questioned.6 That peripheral oedema, frequently observed in advanced COPD patients, reflects true RHF has been debated and even denied, in particular because the degree of pulmonary hypertension is often mild in COPD.15 Peripheral oedema is not synonymous with RHF and may simply indicate the presence of secondary hyperaldosteronism induced by functional renal insufficiency which is, in turn, a consequence of hypercapnic acidosis and/or hypoxaemia.6,15
The role of pressure overload in the development of RHF in these patients has also been debated since the observation that there was no difference in PAP between COPD patients with pronounced peripheral oedema and haemodynamic signs of RHF and other similar patients without oedema and without haemodynamic signs of RHF.16 However, some COPD patients with peripheral oedema do have RHF which is accounted for by a significant worsening of pulmonary hypertension during exacerbations of the disease (table 4).13 In nine of 16 COPD patients with pronounced peripheral oedema, haemodynamic signs of RHF were present during an exacerbation of the disease with a worsening of hypoxaemia and hypercapnia. This worsening, as a result of raised HPV, probably explained the increase in PAP from baseline value and the development of RHF.13
Evolution of arterial blood gases and haemodynamic variables before and during an episode of peripheral oedema in COPD patients
Right ventricular contractility, assessed by the end systolic pressure–volume relation, is generally preserved in respiratory patients with pulmonary hypertension investigated when the disease is in a stable state. The only circumstances where a diminished right ventricular contractility could be documented in COPD patients are in acute exacerbations with presence of peripheral oedema and haemodynamic signs of RHF.16
In summary, many patients with advanced COPD never develop RHF while, on the other hand, at least some patients experience episodes of true RHF during exacerbations of the disease accompanied by a worsening of pulmonary hypertension. Peripheral oedema is not synonymous with RHF and may simply indicate the presence of secondary hyperaldosteronism consequent to respiratory failure.
PROGNOSIS OF COR PULMONALE
The occurrence of documented RHF (peripheral oedema) was classically an indicator of poor prognosis in respiratory patients. In fact it is now accepted that a prolonged survival (≥ 10 years) can be observed after the first episode of peripheral oedema. The prevalence of clinical RHF has greatly decreased with the application of long term oxygen therapy (LTOT), with a resulting improvement in prognosis.
The level of PAP is a good indicator of prognosis in COPD9,17 but also in various categories of chronic respiratory disease such as idiopathic pulmonary fibrosis and sequelae of pulmonary tuberculosis.17 The prognosis is worse in COPD patients with pulmonary hypertension when compared to similar patients without pulmonary hypertension.9 In COPD patients with a mild degree of pulmonary hypertension (20–35 mm Hg) the five year survival rate is about 50%.9,17 The prognosis is particularly poor for patients with severe pulmonary hypertension.17 LTOT greatly improves the survival of hypoxaemic COPD patients18,19 and, accordingly, the prognosis of pulmonary hypertension is improved by LTOT, which could partly be explained by the reduction of RHF episodes with LTOT. Of interest, PAP is still an excellent prognostic indicator in COPD patients receiving LTOT, probably because it is a good marker of both the duration and the severity of alveolar hypoxia in these patients.
TREATMENT OF COR PULMONALE
The treatment of RHF involves diuretics (most often frusemide (furosemide)) and oxygen therapy. Digitalis is used only in the case of an associated left heart failure or in the case of arrhythmia. The treatment of pulmonary hypertension includes vasodilators and LTOT. Is it really necessary to treat pulmonary hypertension in chronic hypoxic lung disease? Is the treatment of the disease itself (for example, COPD) not sufficient? Pulmonary hypertension is generally mild to moderate in COPD and the necessity for treating a mild hypertension can been questioned. An argument in favour of treatment is that pulmonary hypertension, even when modest during a stable period of the disease, may worsen, particularly during acute exacerbations, and these acute increases in PAP can contribute to the development of RHF. The best argument in favour of treatment is that LTOT, which is prescribed to very hypoxaemic respiratory patients, has favourable pulmonary haemodynamic effects.
Chronic cor pulmonale: key points
-
Cor pulmonale can be defined as pulmonary arterial hypertension resulting from diseases affecting the structure and/or function of the lungs. Pulmonary hypertension results in right ventricular enlargement and may lead with time to right heart failure
-
Chronic obstructive pulmonary disease (COPD) is by far the main cause of chronic respiratory insufficiency and cor pulmonale
-
In COPD alveolar hypoxia is the first cause of pulmonary hypertension. Acute hypoxia causes pulmonary vasoconstriction and chronic longstanding hypoxia induces pulmonary vascular remodelling
-
In chronic respiratory disease pulmonary hypertension is of mild to moderate degree, but it may worsen during exercise, sleep, and acute exacerbations of the disease
-
Many patients with advanced COPD will never develop right heart failure, but other patients experience episodes of right heart failure during exacerbations of the disease accompanied by a worsening of pulmonary hypertension
-
Long term oxygen therapy is at present the best treatment for pulmonary hypertension in chronic respiratory failure. Future treatment may combine oxygen therapy and specific vasodilators
Experience with vasodilator therapy has come from the treatment of primary and severe pulmonary hypertension. There are very few selective pulmonary vasodilators. At present, inhaled nitric oxide cannot be administered for long periods because of toxicological reasons. Prostacyclin, bosentan, and sildenafil, which are effective in treating patients with primary pulmonary hypertension, do not seem appropriate for COPD patients and there is, at present, no justification for the long term use of vasodilators in these patients.
One of the aims of LTOT in COPD patients is to attenuate the development of pulmonary hypertension and to reduce the frequency of episodes of RHF. Since alveolar hypoxia is the major determinant of the rise of PVR and PAP, it is logical to treat with LTOT hypoxaemic COPD patients exhibiting pulmonary hypertension. The NOTT (nocturnal oxygen therapy trial)18 and Medical Research Council19 multicentre studies have shown that LTOT improves18 or at least stabilises19 pulmonary hypertension. LTOT has also been shown to reverse the progression of pulmonary hypertension in COPD patients.20 However, PAP rarely returns to normal. The longer the period of LTOT (> 16 hours/day) the better the haemodynamic results. At present LTOT is the best treatment for pulmonary hypertension in COPD patients. In the future, this treatment may combine LTOT and specific vasodilators.