
Key Points
Membranous nephropathy (MN) can be primary/‘idiopathic’ (IMN) or secondary. Identification and eradication of secondary causes is important in all patients
Around 70% of IMN patients will have autoantibodies to PLA2R1 at presentation: these antibodies may be useful in diagnosis and monitoring
All patients with MN should be treated with salt restriction, angiotensin system blockade and modification of cardiovascular risk. In addition, immunosuppressive therapy can benefit selected patients. More RCTs are needed to optimise therapy
Membranous nephropathy (MN) is the most common cause of primary nephrotic syndrome in adults. Recent figures from the Netherlands show an incidence in that country of around 10 per million population per year.1 Identical clinical presentation and histological appearances can occur whether the condition is primary or idiopathic (IMN), or when it is secondary to various drugs, infections or tumours (Table 1). Thus, careful consideration is needed to exclude an underlying cause when assessing a new patient. If one can be identified, the prognosis is that of the underlying condition, management should be directed towards that condition, causative drugs stopped, and infections or tumours treated and eradicated if possible. If successful, the secondary MN can be expected to resolve.
Secondary membranous nephropathy (MN).
Epidemiology
IMN has a slight male preponderance (1.3–2.2:1) and is increasingly common with advancing age, being relatively rare in children in the developed world. The incidence of secondary MN relates to the prevalence of the underlying conditions, in particular infections such as hepatitis B. Consequently, in contrast to IMN, secondary causes of MN are a significant cause of nephrotic syndrome in children in developing countries.2 Although familial cases have been reported, these are perhaps surprisingly rare given the role immunogenetics is now thought to play in the aetiology of the disease.
Several forms of glomerulonephritis are associated with malignancy (see excellent recent review3). MN has the strongest association, with approximately 10% of MN patients having malignancy. This association accounts for 70% of nephrotic syndrome in patients with cancer. The common tumours are shown in Table 1: lung and gastrointestinal carcinomas predominate. Age over 65 and smoking history of over 20 pack-years are substantial risk factors.4 Medications, infections and autoimmune disease are the other important causes of secondary MN.
Natural history
IMN has a variable natural history: about one-third of patients undergo spontaneous remission and have an excellent long-term prognosis, but a similar proportion experience progressive deterioration of excretory renal function, developing chronic kidney disease with all its attendant morbidity and mortality. Some patients have severe, sometimes life-threatening, nephrotic syndrome even when their excretory renal function remains normal. It is important to note that spontaneous remission can be complete or partial but, critically, may take up to two years to occur, with a mean of about one year.5
In view of the variability of the natural history, randomised controlled trials (RCTs) are essential if conclusions about treatment efficacy are to be secure. Aggressive intervention with potentially toxic drugs should be focused on the more severely affected subsets of patients since many patients with IMN will have an excellent prognosis without such treatment. The challenge is to identify prospectively the bad prognosis patients and intervene early before too much damage has been done. This is still not done very well.
Practical points
In every new patient with MN, careful consideration should be given to whether there is an underlying cause such as a drug, infection or tumour.
In view of the variable natural history of IMN, treatment decisions should be based on evidence from RCTs.
Aggressive immunosuppressive treatment should be reserved for patients with progressive loss of excretory kidney function and/or severe nephrotic syndrome.
Pathogenesis
Several significant advances have been made recently in the understanding of pathogenesis and treatment of MN. These will be the focus of this article rather than the older history of the condition which has been extensively reviewed elsewhere.6,7
MN is a discrete pathological entity (Fig 1) which can be identified only by renal biopsy, including examination of the tissue by electron microscopy. The glomerular capillary wall is expanded, but the cellularity of the glomerulus is not typically increased (Fig 1(a)). Immune deposits, typically including immunoglobulin (Ig) G and complement, are found in a granular distribution along the glomerular capillary wall (Fig 1(b)). Electron microscopy shows electron dense deposits in the subepithelial space adjacent to the foot processes of the glomerular epithelial cells or podocytes (Fig 1(c)).
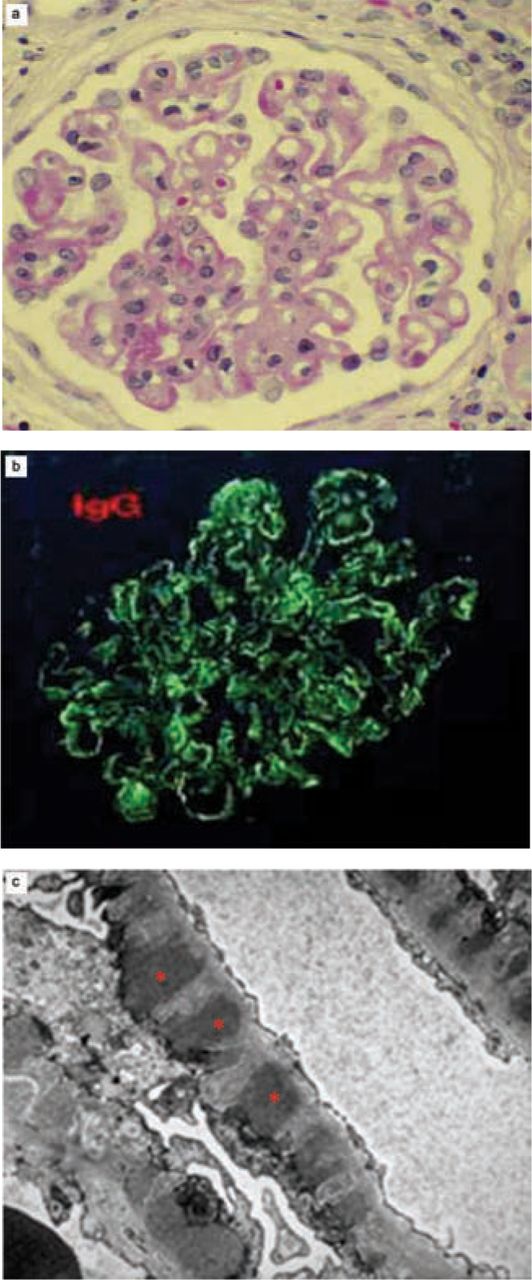
Renal biopsy appearances of membranous nephropathy. (a) Light microscopy (haematoxylin and eosin stain). (b) Immunofluorescence for immunoglobulin G (similar appearances typically seen for C3). (c) Electron microscopy (electron-dense deposits shown by asterisks).
Much of the early understanding of MN was based on analysis of animal models, especially Heymann nephritis in rats.8 It is clear from more recent work that the pathogenesis of IMN in humans is likely to be a conventional autoimmune process in genetically-predisposed individuals, with the podocyte the major target of injury. In fact, an autoimmune pathogenesis has long been assumed in IMN because of the immune deposits in the glomerulus and the analogies with Heymann nephritis. Therapeutic advances (as so often the case) have lagged behind the basic science progress, but there are very good reasons for optimism that the ability to manage IMN will improve rapidly in the next few years.
Recent advances in understanding
Neutral endopeptidase
The first major advance in recent years was the elegant description by Pierre Ronco and colleagues of MN due to alloimmunisation against neutral endopeptidase (NEP). Babies born to NEP-deficient mothers developed transient neonatal MN due to transplacental transfer of anti-NEP induced in the mother by an immune response to NEP in a previous fetus.9 This was a beautiful demonstration of the induction of human MN by an IgG antibody, but unfortunately it soon became clear that NEP was not the auto-antigenic target in sporadic IMN.
Phospholipase A2 receptor
Larry Beck and colleagues reported in 200910 that about 70% of patients with IMN have autoantibodies directed against the phospholipase A2 receptor PLA2R1. This has since been confirmed by others11 and in different populations.12 There is a suggestion that anti-PLA2R1 antibody levels correlate with disease activity and/or with response to treatment.13 Testing for this auto-antibody therefore shows promise, not only in diagnosis of IMN but also in monitoring. Commercial assays for anti-PLA2R1 are now available.
PLA2R1 is expressed by podocytes,14 but it is not yet clear whether the immune response is primarily directed against podocyte PLA2R1 or whether the podocyte is an innocent bystander injured by the anti-PLA2R1 antibody response. Either way, podocytes are the target of injury, presumably mediated by complement. The predominant autoantibodies in IMN are of IgG4 subclass. Although this is not regarded as a complement-activating subclass of IgG, recent data from Beck et al15 show that anti-PLA2R1 autoantibodies activate complement, mainly via the lectin pathway. Novel approaches to therapy could therefore be directed against the anti-PLA2R1 autoimmune response or towards measures to protect, repair and/or regenerate the injured podocytes.
Genetics
The other recent major advance in IMN has been the elucidation of the genetics of the condition. Stanescu et al16 reported genome-wide association studies in three European populations and showed that two genetic regions are closely involved in predisposition to IMN. One is on chromosome 6 in the major histocompatibility complex (MHC), the strongest association being with HLA-DQA1; the other is on chromosome 2, most likely the gene encoding PLA2R1 itself.
It is intriguing that the condition should result from predisposing genes in the MHC, an immune response gene, and in the autoantigen itself, although curiously few familial cases of IMN are reported. The MHC association was the strongest: this, together with the fact that 20–30% of patients with IMN do not apparently have autoantibodies to PLA2R1, suggests that other autoantigens must be involved. Other candidates include aldose reductase and manganese superoxide dismutase.17
Practical points:
About 70% of patients with IMN will have auto-antibodies to PLA2R1. A commercial test for these is available.
Testing for the antibody may come to play a role in diagnosis and monitoring.
Genetic predisposition to IMN (at least in European populations) is tightly associated with two gene loci, one in the MHC and the other probably PLA2R1 itself.
It is surprising that familial cases are not more common.
The male predominance reported in most series is also unexplained by the genetic studies.
Clinical features
As stated earlier, IMN has a variable natural history. About 75% of adults with IMN present with nephrotic syndrome, which can be life-threatening and often with very heavy and sustained proteinuria. The other 25% present with subnephrotic (but still significant) proteinuria, with or without impairment of excretory renal function. Microscopic haematuria is not unusual (40–50%). Renal impairment and hypertension may be present.
Patients may have features suggestive of a secondary cause such as symptoms and signs of underlying malignancy, systemic lupus erythematosus/rheumatoid arthritis or an infective cause (Table 1). For MN associated with malignancy, the two conditions are usually identified within 12 months of each other, 80% of malignancies being present before or at the time of the nephrotic syndrome diagnosis.
The reported incidence of thromboembolism in MN varies hugely in the literature (3–48%) but is undoubtedly a significant risk. A summary of studies suggests 11% of patients have a deep vein thrombosis, 11% a clinically significant pulmonary embolus and 35% (5–60%) renal vein thrombosis (RVT).18 A recent retrospective study demonstrated a hazard ratio of 22 for venous thromboembolism compared to patients with IgA nephropathy. Even after correction for age, cancer history and degree of proteinuria, the hazard ratio was roughly 10 times that of IgA nephropathy and twice as high as focal segmental glomerulosclerosis.19 The ascertainment of RVT rate clearly depends on how aggressively the diagnosis is pursued, but RVT genuinely seems more common in MN and its occurrence carries an adverse renal prognosis. The reasons remain unclear for this difference in thromboembolism and RVT in patients with different pathologies despite similar severity of nephrotic syndrome.
Treatment
Kidney Disease: Improving Global Outcomes (KDIGO) guidelines have recently been published for the treatment of IMN, with graded recommendations on the limited evidence available and offering excellent guidance.20 As in all proteinuric conditions, patients with IMN should receive:
dietary salt restriction
angiotensin system blockade, even if not hypertensive; if they are hypertensive, strict blood pressure control is of course essential
modification of cardiovascular risk by smoking avoidance/lifestyle modification/cholesterol lowering therapy, and
diuretics for oedema control, as required.
There is evidence in favour of prophylactic anticoagulation in patients with severe hypoalbuminaemia due to IMN (≤25 g/l).20 Secondary causes of MN should be screened for and the underlying condition (eg infection or malignancy) treated. It is also important to consider vaccination against encapsulated bacteria and antibiotic prophylaxis in those with persistent hypogammaglobulinaemia.
Immunosuppression
It is more controversial whether patients with IMN should be given immunosuppressive therapy and, if so, how patients should be selected for treatment, when it should be given and which agent(s) used. As already mentioned, data from uncontrolled trials should be treated with great caution because of the variable natural history of the condition. Unfortunately, there is a shortage of controlled trials in IMN. Partly because of spontaneous remission and partly because the risk-benefit studies of immunosuppression in IMN are so limited, the KDIGO guidelines20 recommend initiating immunosuppressive medication only in patients with:
severe, disabling or life-threatening symptoms related to nephrotic syndrome or
persistent nephrotic range (>4 g/l) proteinuria unresponsive to antihypertensive and renin-angiotensin blockade over at least six months or
progressive rise in creatinine (>30%) over 6–12 months unexplained by other renal insults.
It is NOT recommended to initiate immunosuppressive therapy for IMN in patients with small kidneys (eg <8 cm in adults) or if eGFR is below 30 ml/min/1.73m2 or in the presence of severe/life-threatening infection. To prescribe immunosuppressive therapy outside these parameters exposes patients to potentially toxic medications in the absence of any supportive data.
Steroids
There is no evidence that steroids alone or mycophenolate mofetil have any role in the initial treatment of IMN.
A Cochrane review22 and more recently the KDIGO guidelines20 have concluded that the treatment for which there is the best evidence is the combination of prednisolone with an alkylating agent, either chlorambucil or cyclophosphamide. The latter is recommended as first choice because, although remission rates are similar, there may be fewer severe adverse effects.
Ponticelli regimen
This approach was originally popularised from Italy by Claudio Ponticelli.23,24 The Ponticelli regimen consists of a six-month course of treatment with alternating 30-day cycles, starting with 1 g methylprednisolone daily for three days followed by oral methylprednisolone 0.5 mg/kg/day for the rest of the first month. For the second month patients receive either oral cyclophosphamide (2 mg/kg/day) or chlorambucil (0.15–2 mg/kg/day), then repeat the cycle by returning to the steroid regimen.
The 10-year follow-up of the original Ponticelli RCT demonstrated complete or partial remission in 61% of the treated patients compared with 33% of the controls (very few of whom achieved complete remission). The treatment group also had better patient and renal survival.23 In the recently reported UK RCT focusing on the subset of patients with deteriorating renal function, prednisolone plus chlorambucil provided significant protection of renal function compared with cyclosporine or supportive treatment alone.25 Adverse events with this approach are very frequent. For this reason, plus the fact that the deterioration was slowed rather than prevented, better treatments are still needed.
Future treatments
Perhaps the two most promising treatments are rituximab26 and adrenocorticotropic hormone (ACTH).27
Rituximab
Rituximab targets B lymphocytes. Since it is now known that the disease is associated with an auto-antibody, at least in most cases, a drug that prevents synthesis of this antibody seems logical. Rituximab has not yet been tested in RCTs, the responses are mostly in terms of reductions in proteinuria rather than long-term protection of excretory renal function, and there are important concerns about its cost and long-term safety.28
ACTH
Although ACTH has been studied only in small series, it shows promise. It probably works not by stimulating adrenal steroid production but by direct effects on podocytes which express the appropriate receptor and show responsiveness to ACTH in vitro.29
Calcineurin inhibitors
Calcineurin inhibitors (cyclosporine or tacrolimus) are undoubtedly capable of reducing proteinuria, although there is a very high risk of relapse when they are stopped. They should be:
reserved for patients not suitable for or who fail six months of steroids plus an alkylating agent
used with great caution in patients with impaired renal function
reduced to as low a dose as possible in those who respond, and
promptly stopped in those who do not respond.
Practical points
All patients with IMN should be treated with dietary salt restriction, angiotensin cascade inhibitors and/or other antihypertensive agents.
Additional immunosuppression is best reserved for patients with progressive loss of excretory kidney function and/or severe nephrotic syndrome.
The treatment for which there is the best evidence is a six-month course of alternating monthly cycles of high-dose prednisolone and an alkylating agent (chlorambucil or cyclophosphamide).
Novel treatments that show promise in uncontrolled trials include rituximab and ACTH. Well-designed RCTs using these agents are urgently needed.
For secondary MN, there is an interesting report29 that universal vaccination programmes can lead to almost complete eradication of childhood MN secondary to hepatitis B. A similar approach to control other infective agents may well have similar benefits.
Recurrent and de novo membranous nephropathy in renal transplants
Recurrent IMN occurs in 7–44% of patients by three years, up to half of those affected returning to end-stage renal disease (ESRD) by 10 years.30 Recurrent disease presents earlier than de novo MN, typically within two years (15 months vs 4 years for de novo). As yet, there seem to be no markers to identify patients at risk of recurrence, but it may be that titres of anti-PLA2R1 will be predictive.
De novo MN seems to be very common and may be apparent in as many as 2% of all renal transplants. The reason is not clear, but may be associated with donor-specific antibodies (HLA or non-HLA). As with native IMN, some success has been claimed for rituximab in small series,31 but the numbers are small and RCTs are needed before this agent moves into routine use.
Prognosis
The prognosis of secondary MN is highly dependent on the underlying cause and the response to therapy, but there are many examples of remission following treatment of either malignancy or the associated infectious disease. The prognostic indicators for IMN are little different from most other kidney diseases. Adverse factors include:
severity and duration of proteinuria
hypertension
reduced and progressively declining glomerular filtration rate (GFR)
glomerulosclerosis
tubulointerstitial fibrosis/atrophy, and
being male and older age.
Duration and severity of proteinuria are of practical use: 32% of patients with nephrotic range proteinuria develop a decline in GFR compared to 12% of those with subnephrotic proteinuria, and 66% of patients with more than 8 g/d of proteinuria for longer than six months progress to ESRD. However, one-third of patients in this category do not share this fate: approximately 20% of those with very heavy proteinuria undergo spontaneous remission. Conversely, female patients presenting with no tubulointerstitial fibrosis/glomerulosclerosis and who have subnephrotic proteinuria have a good prognosis.
In common with all nephrotic disease, any reduction in proteinuria, in the absence of reduced GFR, is associated with a significantly better outcome: 10-year renal survival is 100%, 90% and 50% in complete, partial and no remission, respectively.
Conclusions
MN is a common cause of nephrotic syndrome in adults and associated with significant morbidity. Distinguishing IMN from secondary MN is critical and anti-PLA2R1 antibodies may help identify IMN. Furthermore, genuinely remarkable developments in the understanding of IMN are afoot which will hopefully result in better treatments. Well-designed RCTs of newer agents are urgently needed.
The National Kidney Federation has a good website providing patient information. The membranous page is a good first stop for newly diagnosed patients.32
- © 2012 Royal College of Physicians
References








{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.